








This review article covers ketamine’s efficacy in psychiatric conditions, including depression, bipolar disorder, anxiety, PTSD, and OCD. It also includes information on its mechanism, safety, comparisons to other novel and relevant antidepressants, and background information on both ketamine and the disorders it is used for.
You can download a PDF of the review article here.
Contents
- Section 1: Introduction
- Section 2: Background
- Section 3: Introduction to Ketamine
- Section 4: Racemic Ketamine For Mood Disorders
- Section 5: Ketamine For Other Psychiatric Disorders
- Section 6: Biomarkers, Predictors, and Correlates
- Section 7: Antidepressant Mechanism
- 7.1 Glutamatergic Activity
- 7.2 mTOR
- 7.3 Monoamines
- 7.4 S-Ketamine or R-Ketamine?
- 7.5 BDNF-TrkB Signaling
- 7.6 GSK-3
- 7.7 Modulation of Inflammation and Immune System Activity
- 7.8 VEGF
- 7.9 Opioid Receptors
- 7.10 Gut Microbiome
- 7.11 Sex Differences
- 7.12 Regions of Interest
- 7.13 HCN Channels
- 7.14 microRNA
- 7.15 HDAC
- 7.16 Other
- Section 8: The Role of Metabolism
- Section 9: Risks
- Section 10: Comparison With Other Drugs
- References
Section 1: Introduction
1.1 Overview
Mood disorders like major depressive disorder (MDD) and bipolar disorder (BD) are leading contributors to disability, yet the efficacy of treatments for those conditions has barely changed in more than half a century. Most people either do not respond to standard treatments, do not adequately respond, or they lose their response shortly after stopping. Therefore, there is a large burden of treatment-resistant depression (TRD) and poorly treated depression.
Following research in the 1990s that suggested the glutamate system may be a shared downstream target of antidepressants ranging from selective serotonin reuptake inhibitors (SSRIs) to tricyclic antidepressants (TCAs) and electroconvulsive therapy (ECT), some researchers began investigating drugs that more directly affect that system. This led to the discovery that inhibitors of the glutamatergic NMDA receptor (NMDAR) have antidepressant-relevant effects in animals and can reverse stress- and depression-related neurological changes.
Soon thereafter, researchers at Yale University published the first double-blind randomized controlled trial (DBRCT) of ketamine for depression. They found a single infusion rapidly alleviated depressive symptoms and worked for at least three days. Dozens of studies and case reports have been published on ketamine’s effect in psychiatric conditions since that initial trial. Efficacy has been reported in MDD, BD, posttraumatic stress disorder (PTSD), anxiety, obsessive-compulsive disorder (OCD), and substance use disorders.
It usually reduces depressive symptoms for at least three days followed by a return to baseline within a week, though the duration can be extended to a couple weeks if multiple infusions are used. The short duration of efficacy per dose is far from ideal since ketamine is typically only available at clinics and other medical facilities, treatment can be expensive, and there are concerns about the safety of repeated dosing. However, it is effective in people who have failed to respond to standard treatments and augmentation therapies, so it is helpful despite its downsides.
Ketamine may also be particularly useful in a few targeted situations. First, multiple studies have reported an anti-suicidal effect, making it one of the only interventions for people who are acutely suicidal and could be at risk of self-harm if they were to wait weeks to respond to common treatments. Second, when ketamine is given at the start of therapy with standard medications like SSRIs, it can reduce how long it takes for people to benefit. And third, it may alleviate anhedonia (reduced capacity to feel pleasure and/or impaired motivation to seek out pleasure), a symptom that can be relatively resistant to common treatments.
Those benefits seem to involve some of the same mechanisms that have been implicated in the effect of existing antidepressants, which can take weeks to really alleviate depression, indicating their initial mechanism—usually inhibiting the reuptake or breakdown of monoamine neurotransmitters like serotonin and norepinephrine—is not directly responsible for their effect. Researchers have known for decades that the basic monoamine or serotonin theory of depression, which suggests depression comes from a ‘deficit’ in monoamines, is misleading at best. Instead, depression physiology and antidepressant mechanism likely have a larger connection to neuroplasticity, including changes in synaptogenesis (creation of new synaptic connections between neurons) and synaptic plasticity (altered strength of those connections). Neurogenesis, i.e. the generation of new neurons, might be involved to some extent in a mood-relevant brain region like the hippocampus, but neuroplasticity seems to be more important.
Common treatments take days or weeks to produce those changes, whereas ketamine works almost immediately. Soon after administration it upregulates synaptic protein synthesis and increases synaptic connections, particularly in the prefrontal cortex (PFC) and hippocampus, facilitating neuroplasticity.
Clinical studies rely on formal measurements of psychiatric symptoms and treatment side effects, so those measurements are detailed in Section 1.2, and because most of the research on ketamine’s mechanism is conducted in animals, Section 1.3 discusses some of the major animal models of depression and how antidepressant activity is tested outside of humans.
Section 2 provides background information on what we know about the pathophysiology of depression, the role of glutamate and neuroplasticity, and why new treatments are needed. Ketamine’s pharmacology, general effects, and history are introduced in Section 3.
The effect of ketamine on depression, bipolar disorder, PTSD, substance use, and other conditions is detailed in Sections 4 and 5. Correlates and predictors of efficacy, such as neurotrophin levels and family history, are discussed in Section 6. The proposed mechanisms of action are found in Section 7, which also details what we know about the specific roles of S-ketamine and R-ketamine.
Since the publication of a paper suggesting metabolism to (2R,6R)-hydroxynorketamine is a major contributor to the effects of ketamine, metabolism has been studied by several researchers; findings from those papers are found in Section 8. Ketamine’s acute and chronic risks are discussed in Section 9.
Lastly, the therapeutic potential of ketamine has led to research on other glutamatergic drugs, most of which have failed to replicate ketamine’s effects. Section 10 discusses results from research on those substances, GABAergic drugs, and the muscarinic acetylcholine receptor antagonist scopolamine, which appears to be a rapid-acting antidepressant with a mechanism similar to that of ketamine.
1.2 Human Research Methods
Depression
Hamilton Depression Rating Scale (HDRS; HAM-D)
The HDRS is a depressive symptom questionnaire commonly administered by clinicians (Hamilton, 1960; Sharp, 2015). In its original form, which was created in the late 1950s to measure antidepressant efficacy, the questionnaire contained 17 items (HDRS-17). The HDRS-17 may be referred to as the HDRS-21 because it contains four additional items pertaining to depression subtype and uncommon symptoms, but those items do not contribute to scoring. Some items are rated 0-2 and others are rated 0-4.
Other versions have been developed with different numbers of items, with the HDRS-24 also being common.
The HDRS has been a standard assessment tool for decades, but it has been criticized for failing to capture key symptoms like anhedonia and feelings of worthlessness (Sharp, 2015).
Scoring
HDRS-17
- Normal: 0 – 7
- Mild depression: 8 – 16
- Moderate depression: 17 – 23
- Severe depression: >24
Items
The original 17 items were: (1) depressed mood; (2) guilt; (3) suicidality; (4-6) initial, middle, or delayed insomnia; (7) work and interests (e.g. feelings of incapacity, loss of interest in hobbies, reduced productivity); (8) retardation (e.g. apathy, slowed speech or activity); (9) agitation; (10-11) anxiety-psychic and anxiety-somatic; (12-13) gastrointestinal (e.g. appetite loss, constipation) and general somatic symptoms (e.g. fatigue, heaviness in limbs or head); (14) genital symptoms (e.g. loss of libido); (15) hypochondriasis; (16) loss of insight; and (17) weight loss.
Hamilton (1960) included four additional unscored items in the original version: diurnal variation, depersonalization, paranoid symptoms, and obsessional symptoms.
Montgomery–Åsberg Depression Rating Scale (MADRS)
The MADRS is a 10-item questionnaire for measuring depressive symptoms (Montgomery and Asberg, 1979). It was developed in the 1970s to improve the observation of antidepressant effects because existing assessment tools inadequately differentiated active treatments from each other despite effectively distinguishing active drugs from placebo. Scores on this measure strongly correlate with the HDRS.
The 10 MADRS items were chosen based on which components of the 65-item Comprehensive Psychopathological Rating Scale (CRPS) most accutely captured depressive disorders. Each item is scored on a 0-6 scale.
Scoring
Different cutoff points for depression severity have been used by different research groups. A common scoring guide is:
- Normal: 0 – 6
- Mild depression: 7 – 19
- Moderate depression: 20 – 34
- Severe depression: >35
Items
The 10 items are: (1) apparent sadness; (2) reported sadness; (3) inner tension (e.g. ill-defined discomfort, edginess, tension leading to panic or dread); (4) reduced sleep; (5) reduced appetite; (6) concentration difficulties; (7) lassitude (e.g. slowness initiating and performing everyday activities); (8) inability to feel; (9) pessimistic thoughts; and (10) suicidal thoughts.
Beck Depression Inventory (BDI)
The first version of the BDI was published in 1961 and it remains in use as a depression measurement tool (Beck et al., 1961). It was developed based on clinical observation of the symptoms frequently reported by depressed patients that were deemed relatively specific to depressive disorders. The BDI contains 21 items scored on a 0 to 3 scale.
Scoring
No specific cutoff scores were outlined for the BDI because different scoring was recommended depending on the application (Beck and, 1974). For screening purposes, cutoff points of 10 or 13 were found to be appropriate, whereas a higher cutoff of >20 was recommended for identifying a ‘pure’ group of depressed patients and minimizing false positives in research settings. The following guideline was commonly used in clinical situations (Beck et al., 1988):
- Normal: 0 – 9
- Mild depression: 10 – 18
- Moderate depression: 19 – 29
- Severe depression: 30 – 63
Items
In the original BDI, the 21 items were: (1) mood; (2) pessimism; (3) sense of failure; (4) lack of satisfaction; (5) guilty feeling; (6) sense of punishment; (7) self-hate; (8) self-accusations; (9) self-punitive wishes; (10) crying spells; (11) irritability; (12) social withdrawal; (13) indecisiveness; (14) body image; (15) work inhibition; (16) sleep disturbance; (17) fatigue; (18) appetite loss; (19) weight loss; (20) somatic preoccupation; and (21) loss of libido.
Suicidality
Scale for Suicide Ideation (SSI)
Beck et al. (1979) created the clinician-administered SSI to measure the magnitude of current suicidal ideation. It includes 19 items scored on a 0 to 2 scale.
Different cutoffs for suicidality have been used, including ≥4 and ≥6 (Holi et al., 2005; de Beurs et al., 2016).
Items
The 19 items of the SSI are: (1) wish to live; (2) wish to die; (3) reasons for living/dying (i.e. more reasons for living or dying); (4) desire to make active suicide attempt; (5) passive suicidal desire; (6) duration of suicide ideation/wish; (7) frequency of suicidal ideation; (8) attitude toward ideation/wish; (9) degree of control over acting out suicidal action; (10) concern for deterrents to active attempt (e.g. family, religion); (11) reason for contemplated attempt (e.g. revenge, attention, escape); (12) specificity of contemplated attempt; (13) availability/opportunity for contemplated attempt; (14) sense of ‘capability’ to carry out the attempt; (15) expectancy/anticipation of actual attempt; (16) actual preparation for contemplated attempt; (17) suicide note; (18) final acts in anticipation of death (e.g. insurance, will); and (19) deception/concealment of contemplated suicide.
Anxiety
Hamilton Anxiety Rating Scale (HAM-A)
Published in 1959, the HAM-A was one of the earliest clinical assessment tools for anxiety (Hamilton, 1959). It was developed to measure anxiety symptoms in patients with a preexisting ‘neurotic anxiety’ diagnosis rather than for measuring anxiety in other clinical populations. The HAM-A includes 14 items scored on a 0 to 4 scale.
Scoring
Matza et al. (2010) recommended the following scoring guideline based on a study of 144 generalized anxiety disorder patients:
- Normal: 0 – 7
- Mild anxiety: 8 – 14
- Moderate anxiety: 15 – 23
- Severe anxiety: ≥24
Items
The 14 items are: (1) anxious mood (e.g. worries, apprehension); (2) tension (e.g. unable to relax, feeling of tension); (3) fears (e.g. of dark, traffic, crowds); (4) insomnia; (5) cognitive (e.g. poor memory or concentration); (6) depressed mood; (7) somatic-muscular (e.g. muscle tension, unsteady voice, teeth grinding); (8) somatic-sensory (e.g. tinnitus, hot and cold flushes); (9) cardiovascular (e.g. tachycardia, palpitations); (10) respiratory (e.g. chest constriction, dyspnea); (11) gastrointestinal (e.g. weight loss, dyspepsia); (12) genitourinary (e.g. urinary frequency or urgency, erectile dysfunction, amenorrhea); (13) autonomic (e.g. dry mouth, sweating, tension headache); and (14) behavior at interview (e.g. unrelaxed, fidgeting).
PTSD
Clinician-Administered PTSD Scale (CAPS)
The CAPS was developed in 1989 at the National Center for PTSD for use among clinicians and researchers (Blake et al., 1995). It measured 30 items on separate 0 to 4 scales for frequency and intensity, specifically focusing on current PTSD symptoms experienced within the past month.
To correspond with the diagnostic criteria used in the DSM-5, the CAPS for DSM-5 (CAPS-5) was created (Weathers et al., 2015, 2018). It consists of 30 items covering the 20 DSM-5 PTSD symptoms, subjective distress, functional impact, severity, onset, and duration. Unlike earlier versions, the CAPS-5 measures each item with a single 0 to 4 severity score, not separate frequency and intensity scores. There are three main versions of the scale: past-week, past-month, and worst-month (lifetime PTSD). Diagnosis is made using the past-month version.
For PTSD diagnosis, the CAPS-5 requires the existence of an index traumatic event (Criterion A), at least one intrusion symptom (Criterion B), at least one avoidance symptom (Criterion C), at least one negative change in cognition and/or mood associated with the traumatic event (Criterion D), and at least one symptom of altered arousal and reactivity (Criterion E). Additionally, those symptoms must have a duration of ≥1 month (Criterion F) and be associated with distress or impairment (Criterion G).
Psychosis
Brief Psychiatric Rating Scale (BPRS)
The earliest version of the BPRS was published in 1962 and included 16 items relevant to psychotic disorders, depression, anxiety, and other psychiatric conditions, though it has been particularly popular as a measurement tool for symptoms of psychosis (Overall and Gorham, 1962). Each item is scored on a 1-7 scale.
Positive psychotic symptoms have been measured using the suspiciousness, hallucinations, unusual thought content, and conceptual disorganization subscales, while negative psychotic symptoms have been measured with the blunted affect, emotional withdrawal, and motor retardation subscales.
Items
The 16 items in the original BPRS were: (1) somatic concern (e.g. concerns about bodily health); (2) anxiety; (3) emotional withdrawal; (4) conceptual disorganization; (5) guilt feelings; (6) tension; (7) mannerisms and posturing (e.g. unusual and unnatural motor behavior); (8) grandiosity; (9) depressive mood; (10) hostility; (11) suspiciousness; (12) hallucinations; (13) motor retardation; (14) uncooperativeness; (15) unusual thought content; and (16) blunted affect.
Dissociative States
Clinician-Administered Dissociative States Scale (CADSS)
Bremner and colleagues (1998) developed the CADSS based on a review of the relevant literature and they tested it on patients with combat-related PTSD and significant dissociative symptoms. The CADSS contains 27 items formatted as questions, which are either answered by the subject (items 1-19) or the observer (items 20-27). Each item is scored on a 0 to 4 scale.
Scoring
No scoring guideline was recommended by Bremner et al. (1998) and the CADSS has not been used as much as scales like the HDRS and HAM-A, so less is known about the meaning of specific total scores and subscale scores. In the original paper, PTSD patients with dissociative symptoms had an average score of 19, compared with 3.7 for patients with schizophrenia, 7.5 for patients with affective disorders, 1.5 for healthy controls, and 1.3 for Vietnam combat veterans without PTSD.
Items
The subject-rated items include:
| (1) Do things seem to be moving in slow motion? (2) Do things seem unreal, as if you are in a dream? (3) Do you have some experience that separates you from what is happening; for instance, do you feel as if you are in a movie or a play, or as if you are a robot? (4) Do you feel as if you are looking at things from outside of your body? (5) Do you feel as if you are watching the situation as an observer or spectator? (6) Do you feel disconnected from your body? (7) Does your sense of your own body feel changed: for instance, does your own body feel unusually large or small? (8) Do people seem motionless, dead, or mechanical? (9) Do objects look different than you would expect? (10) Do colors seem to be diminished in intensity? | (11) Do you see things as if you were in a tunnel, or looking through a wide-angle photographic lens? (12) Does this experience seem to take much longer than you would have expected? (13) Do things seem to be happening very quickly, as if there is a lifetime in a moment? (14) Do things happen that you later cannot account for? (15) Do you space out or in some way lose track of what is going on? (16) Do sounds almost disappear or become much stronger than you would have expected? (17) Do things seem to be very real, as if there is a special sense of clarity? (18) Does it seem as if you are looking at the world through a fog, so that people and objects appear far away or unclear? (19) Do colors seem much brighter than you would have expected?
|
The observer-rated items include:
| (20) Did the subject seem eerie or strange, or in some other way give you an uncomfortable feeling? (21) Did the subject blank out or space out, or in some other way appear to have lost track of what was going on? (22) Did the subject appear to be separated or detached from what is going on, as if not a part of the experience or not responding in a way you would expect? | (23) Did the subject say something bizarre or out of context, or not speak when you would have expected it? (24) Did the subject behave in a bizarre, unexpected manner, or show no movement at all, being stiff and wooden? (25) Did the subject have to be put back on track, or grounded in the here and now, during or soon after the experience? (26) Did the subject show any unusual twitching or grimacing of the face? (27) Did the subject show any unusual rolling of the eyes upward or fluttering of the eyelids? |
Mania
Young Mania Rating Scale (YMRS)
The YMRS, a clinician-administered questionnaire for assessing the severity of mania, was introduced in 1978. It includes 11 items scored on a 5-point scale; items 5, 6, 8, and 9 receive twice the weight to compensate for poor cooperation in severely ill patients, resulting in a 0-60 scale (Young et al., 1978). The items are based on the core symptoms of mania associated with bipolar disorder.
Scoring
A common cutoff for inclusion in RCTs pertaining to mania is ≥20, while a cutoff of ≥13 has been used to detect at least mild manic symptoms in bipolar disorder (BD) patients (Busk et al., 2020). Lukasiewicz et al. (2013) reported an average baseline YMRS score of 26.4 in a study of 3255 BD patients with acute mania/mixed state.
Items
The 11 items on the YMRS are: (1) elevated mood; (2) increased motor activity-energy (e.g. hyperactivity, animated); (3) sexual interest (e.g. hypersexuality, overt sexual acts); (4) sleep reduction; (5) irritability; (6) altered speech rate and amount (e.g. talkative, pressured speech); (7) language-thought disorder (e.g. distractible, flight of ideas, incoherent); (8) thought content (e.g. grandiose, delusions, questionable plans); (9) disruptive-aggressive behavior (e.g. demanding, threats, assaultive); (10) appearance (e.g. poorly groomed, disheveled); (11) insight (e.g. ranging from admitting illness or denying behavior change).
1.3 Animal Research Methods
Models of Depression
Chronic Mild Stress (CMS; CUMS)
The chronic mild stress (CMS) model of depression, also known as the chronic unpredictable mild stress (CUMS) model, is a standard way of inducing depressive-like behavior in rats and mice (Willner, 2017). It is considered by many researchers to be one of the best animal models for studying depression and antidepressant action. Repeated stress exposure has been known to cause behavioral changes in rodents since the 1980s, including reduced preference for sucrose-sweetened water, which may be reminiscent of anhedonia in humans. Importantly, chronic treatment with common antidepressants can reverse CMS-related behavioral changes.
While the original methodology involved severe stressors (e.g. intense footshocks or prolonged periods without food and water), ethical concerns led to the creation of a procedure based around comparatively ‘mild’ stressors (Willner, 2017). The idea that CMS is in some way modelling symptoms of human depression is plausible considering the recognized role of chronic stress in depression, but it is difficult to know if behavioral changes that look like symptoms of depression are qualitatively and neurologically the same. Neurobiological research has demonstrated CMS-related changes in monoaminergic and neurotrophin systems that parallel some of the changes associated with human depression, providing some additional support for the model (Hill et al., 2012).
Protocol
The exact protocol varies, but the general design involves ~4 to 6 weeks of daily stressor application in a random order or at weekly intervals; often there are two or three stressors each day. Some of the common stressors are repeated reversal of light/dark cycle, illumination during the night, damp or cold bedding, intermittent bells or white noise, space confinement, 45° cage tilt, food deprivation, and strobe lights. The duration of each stressor ranges from 1-12 hours (Elizalde et al., 2010; Autry et al., 2011; Bessa et al., 2009).
Chronic Social Defeat Stress (CSDS)
The CSDS model (aka resident-intruder test) uses social stress to change affective and social behavior in rodents, most often mice (Golden et al., 2011; Hollis and Kabbaj, 2014). In this paradigm, one animal (the ‘intruder,’ e.g. a male C57BL/6J mouse) is repeatedly exposed to a larger animal prescreened for aggressiveness (‘the resident,’ e.g. a male CD-1 mouse). Some mice are largely unaffected, but most develop anhedonic-like, social avoidance, and anxiety-like behavior within 10 days of CSDS; some effects can be seen after 1-4 days of social defeat.
Although female mice have occasionally been used, the bulk of the research is on male mice, exploiting the aggressive behavior displayed by those animals. Chronic treatment with standard antidepressants can reverse CSDS-induced behavior change.
Protocol
A common procedure for the CSDS is to place an intruder C57BL/6J mouse into the home cage of a different aggressive CD-1 resident for 5-10 min each day for 10 days; different residents are used to avoid habituation (Golden et al., 2011; Qu et al., 2017). The resident attacks the intruder, causing it to adopt a submissive position and freezing behavior. The intruder may be wounded during the attack, but wounding is not required. After 5-10 min of interaction directly within the resident’s home cage compartment, the intruder spends the rest of the 24 h period on the other side of the cage separated by a plexiglass barrier to maintain sensory contact with the resident while being physically separate; therefore, psychological stress persists beyond the 5-10 min daily session.
Intruder mice are housed on their own for one day after the final CSDS session and then a social interaction test (SIT) is performed. During the SIT, the intruder is placed in an arena containing an ‘interaction zone’ near a new aggressive resident mouse housed in a wire-mesh cage. The amount of time spent in the interaction zone determines whether a mouse is susceptible or resilient to social defeat; susceptible mice spend more time avoiding the interaction zone. 70-80% of mice will usually be susceptible.
Learned Helplessness (LH)
Like the CMS model, the LH model affects behavior by exposing animals to an uncontrollable stressor, specifically inescapable shocks. Animals without that experience learn to avoid escapable shocks after the first exposure, but deficits in escape and avoidance behavior are observed in animals previously exposed to inescapable shocks.
Early research on this behavior was performed in dogs in the late 1960s and early 1970s. Seligman (1972) reported that naïve dogs quickly moved to escape a shock and learned to avoid the shock area entirely within a few sessions. In contrast, dogs previously exposed to inescapable shocks became immobile and passively endured the escapable shock until it was over, and they continued to show escape deficits in subsequent sessions. A similar procedure is used for rats and mice (Vollmayr and Gass, 2013; Koike et al., 2011). Escape occurs with movement across a shuttle box from shock to no-shock compartments, with a lever-press, or with wheel-turning. The ideal escape option is somewhat species-specific, as shuttle boxes are preferable to the other options in mice.
Protocol
In the shuttle-box setup, rodents are exposed to inescapable shocks for 1-2 sessions and then escape/avoidance behavior is studied during escapable shock sessions for a few days. Animals are divided into ‘helpless’ and ‘non-helpless’ groups based on latency to escape and percentage of escape failures (e.g. not escaping the shock within 10 sec).
Measures of Antidepressant Activity
Forced Swim Test (FST)
First described in the 1970s, the FST (aka Porsolt test) is a popular behavioral tool for rapidly identifying putative antidepressants. Porsolt et al (1978) described FST-induced behavior in mice and rats as “resembling depression,” although we now know that is a poor way to understand the test. Rodents are forced to swim in a cylinder where escape is impossible, causing them to become immobile after initially exhibiting substantial escape behavior like swimming and climbing.
To this day, immobility is often framed as ‘depressive behavior’ caused by learned helplessness, but unlike in human depression or in subacute models of depression in animals, the behavior change is immediate and may only reflect acute coping behavior (Molendijk and Kloet, 2019). Animals can cope with a stressor actively or passively, and the latter response is not the same as depression.
The FST works for antidepressant drug screening and various antidepressants (including TCAs, MAOIs, SSRIs, and bupropion) are active in it (Kara et al., 2018). While this is useful for detecting putative antidepressants with an easily performed test, the acute efficacy of drugs that require chronic administration to be effective against most symptoms in humans or in depression models like the CMS suggests the underlying mechanism may not map onto core depression symptoms. Therefore, although I refer to FST immobility as ‘depressive behavior’ throughout this review when that is how the test was used, this caveat must be kept in mind. Reduced immobility can be a sign of antidepressant activity, but immobility itself should not be viewed as the rodent equivalent of human depression symptoms.
To reduce false positives and negatives, locomotion should be measured outside the FST. Locomotor stimulants can be anti-immobile and sedatives can be pro-immobile in ways that are not reflective of potential antidepressant-like activity (Slattery and Cryan, 2012).
Protocol
Animals are placed in a swim cylinder with (rats) or without (mice) a pretest swim 24 h before the test session (Slattery and Cryan, 2012; Kara et al., 2018). Immobility is recorded during the last 4 min of a 6-min swimming period, with immobility classified as passive floating except for movements required for the animal to keep its head above water. Swimming (horizontal movement through the cylinder) and climbing (upward movement, usually against the side of the cylinder) may also be recorded.
Tail Suspension Test (TST)
The TST is like the FST in that it also measures acute escape-related behavior during an uncontrollable stressor. It was first detailed by Steru and colleagues in the 1980s, who reported that antidepressants (e.g. amitriptyline, imipramine, mianserin) reduced immobility when mice were suspended by the tail and that an anti-immobility effect in the absence of general locomotor stimulation distinguished antidepressants from other drugs, like amphetamine, diazepam, and antipsychotics (Steru et al., 1985, 1987).
This test is subject to the same caveat as the FST, namely that immobility reflects acute coping behavior, not depressive-like behavior. As with the FST, antidepressants can be acutely effective even if they require chronic administration for most of their efficacy in humans and in animal models like CMS. An anti-immobility effect from a drug that does not increase general locomotion can be indicative of antidepressant potential, but immobility itself is not the same as depressive behavior.
Because the TST can be painful for rats and heavy mice, it is best used in normal-weight mice (Can et al., 2012)
Protocol
Mice are suspended by the tail using tape and a suspension bar (Can et al., 2012). Activity is recorded for 6 minutes and immobility during the entire period is measured. Mice are considered immobile when they are passive and motionless, while mobility includes attempts to reach the suspension bar or the walls of the suspension box, strong shaking, and running-like movements.
Novelty-Suppressed Feeding Test (NSFT)
Reduced feeding in a novel environment, known as hyponeophagia, is measured in the NSFT. It most directly measures anxiety-related behavior, but it is also used to screen for antidepressants in mice and rats (Samuels and Hen, 2011; Blasco-Serra et al., 2017). Unlike tests that are more specific to depression-related behavior, classic anxiolytics are active in the NSFT, including benzodiazepines and barbiturates. Common antidepressants, including SSRIs and TCAs, are active specifically with chronic administration. The utility of this test in antidepressant screening may be connected to the high prevalence of anxiety among people with depression and partially shared neurobiological causes of anxiety- and depression-vulnerability.
Protocol
Animals are deprived of food for 24 h and then placed in a brightly lit open-field arena, with food located in the center of the arena. Latency to eat is recorded during a 5-10 min testing period. Appetite can be controlled for by measuring latency to eat and the amount of food eaten in the animal’s home cage during a certain amount of time (e.g. 5 min).
Sucrose Preference Test (SPT)
Anhedonia, an impairment in the ability to experience pleasure and/or the drive to seek out pleasure, is common in depression. It has been studied in animals by assessing changes in the response to rewarding stimuli after chronic stress exposure. Those changes include deficits in the rewarding properties of drugs in the place preference procedure and deficits in the preference for sucrose-sweetened water over regular water, which is measured in the SPT; alternatively, some studies measure consumption of sugary food. The SPT is performed in mice and rats (Liu et al., 2018; Toth et al., 2008).
Baseline sucrose preference varies by rodent strain, though Pothion et al. (2004) found a sucrose solution was preferred to water in the 11 strains of mice they studied, just to varying degrees. Three strains were exposed to seven weeks of CMS, which significantly reduced sucrose preference in the early weeks, while the deficit resolved by the final week.
Protocol
As an example of the protocol used for the SPT in conjunction with stress, Liu et al. (2018) detailed their methodology for studying the effect of 28 days of CMS on sucrose preference. Before the first test day, mice were adapted to bottles containing sucrose (1% wt/vol) and water, as well as to the SPT apparatus. A baseline reading was taken, then the mice were deprived of food and water for 24 h, followed by the preference test. The SPT was performed again during the last week of the CMS procedure, which was demonstrated to reduce sucrose preference.
Section 2: Background
Mood disorders like major depressive disorder (MDD), bipolar depression, and dysthymia are common mental health issues with an overall past-year prevalence of ~10%, (Kessler et al., 2005). Using data from the Global Burden of Disease 2010 Study, which analyzed the epidemiology and impact of 291 diseases and injuries, Murray (2013) reported two of the top five diseases in the US with the greatest number of years lived with disability (YLDs) were MDD and anxiety disorders. MDD ranked fifth for overall disease burden assessed by disability-adjusted life years (DALYs), i.e. the number of years lost due to mortality or disability, with only ischemic heart disease, chronic obstructive pulmonary disorder (COPD), low back pain, and lung cancer surpassing it; and 10 of the leading causes of DALYs (including MDD) increased more than 30% between 1990 and 2010 (Murray, 2013).
Sex Differences
Depression appears to be more common and more severe in females than in males. Among 1500 MDD patients studied in the STAR*D trial, more females had previously attempted suicide (20% vs. 14%) and depression severity was greater, although certain aspects of psychopathology were more common in males (Marcus et al., 2005). The prevalence of general anxiety disorder (GAD), somatoform disorders (i.e. psychiatric conditions causing physical symptoms), and bulimia was higher in females, but males had a higher prevalence of OCD, alcohol abuse, and drug abuse. Symptomatic differences included higher rates of appetite increase, weight increase, low energy, somatic symptoms, sympathetic arousal, gastrointestinal (GI) symptoms, and interpersonal sensitivity among females, whereas males experienced greater weight decrease and psychomotor agitation.
The apparent distinction between the sexes develops through adolescence and into adulthood. Depression prevalence is similar before puberty, but later in life the prevalence is around twice as high in females (Grigoriadis and Robinson, 2007). Consistent with the STAR*D data, Grigoriadis and Robinson reported females were 2-4x more likely to have a seasonal aspect to their disorder, atypical features (i.e. psychomotor retardation, increased appetite, weight gain), somatic symptoms, rumination, and feelings of worthlessness and guilt.
2.1 Impact of Depression
Suicide
General mortality and suicide were substantially elevated in unipolar depression (n=39,182) and BD (n=15,386) patients in Sweden (Osby et al., 2001). The standardized mortality ratio (SMR), i.e. the ratio of mortality compared to the age-specific average, was 15 and 22 for males and females with BD, respectively, and in unipolar depression, the SMR was 21 for males and 27 for females.
Some antidepressants may increase the risk of suicidal behavior early in treatment, particularly in the first nine days, based on a study of 160,000 patients prescribed amitriptyline, fluoxetine, paroxetine, or dothiepin (a TCA) (Jick et al., 2004). Compared to dothiepin, the relative risk (RR) for nonfatal suicidal behavior was 0.83 for amitriptyline, 1.16 for fluoxetine, and 1.29 for paroxetine, and the RR in patients who were first prescribed an antidepressant within nine days of their suicidal ideation or behavior was 38.0 compared to those who were prescribed an antidepressant ≥90 days before the onset of suicidal symptoms.
An analysis of CDC data on violent deaths in 16 US states identified 16,000 fatal incidents, 61% of which were suicides (Karch et al., 2012). Suicide typically occurred in the context of mental health, intimate partner, or physical health issues, or shortly after a recent crisis. 41% of suicide victims were tested for antidepressants, with a positive rate of 23% (n=947). This serves as weak evidence that antidepressants do not adequately prevent suicide and other interventions (drugs or otherwise) are needed. 44% of suicide victims had a diagnosed psychiatric disorder and only 31% were receiving treatment, based on the 90% of cases in which the precipitating circumstances were known. 74% of people with a diagnosis had depression, 15% had BD, and 11% had an anxiety disorder.
Despite increased access and promotion of mental healthcare, as well as further development in psychiatric medicine, suicide rates have increased in the US since the late 1990s following a period of decline from 1986 to 1999 (Curtin et al., 2016). Particularly among adolescents and young adults, for whom suicide is already a leading cause of death, the rate of suicide increased from 1999 through 2014; the rate also increased for middle-aged adults. Compared to 1999, the overall age-adjusted suicide rate in 2014 increased from 10.5 to 13 per 100,000 (+24%). The rate for females increased from 4.0 to 5.7 and the rate increased from 17.8 to 20.7 for males.
Functioning
The alleviation of depression is associated with better functional outcomes (e.g. work status) and potentially lower healthcare costs. Simon et al. (2000) evaluated the outcome of MDD patients (n=290) initiated on an antidepressant (desipramine, fluoxetine, or imipramine) and found 41% were remitted, 47% were improved, and 12% were persistently depressed at 12 months. Patients with greater depression reduction over time were more likely to maintain paid employment and they reported fewer days missed from work because of illness, after adjusting for baseline comorbidities and depression severity. Superior 12-month outcome had a trend-level correlation with reduced healthcare costs in the second year of follow-up.
2.2 Neurological Correlates
Clinical and preclinical research has shown depression is associated with a reduction in brain size in areas relevant to mood and cognitive function, such as the PFC and hippocampus, and those areas exhibit impair synaptic connections between neurons (Duman et al, 2012).
Human Research
Compared to healthy controls (n=10), patients (n=10) with a history of major depression who were in remission had smaller left and right hippocampal gray matter volume, but they did not differ in total cerebral volume (Sheline et al., 1996). The extent of hippocampal volume deficit in the patient group correlated with the total duration of their depression. Rajkowska et al. (1999) observed alterations in the prefrontal cortex (PFC) of postmortem samples from depressed (n=12) compared to control (n=12) individuals. Those with a history of depression had reduced cortical thickness, neuronal size, and neuronal and glia density in the upper (II-V) cortical layers of the rostral orbitofrontal cortex (OFC). Glia density and neuronal size were also reduced in the lower (V-VI) layers of the caudal OFC. In the supra- and infragranular layers of the dorsolateral PFC (dlPFC), depressed subjects had a reduction in the density and size of neurons and glia.
A study comparing postmortem brain tissue from MDD (n=15), BD (n=15), schizophrenia (n=15), and control (n=15) individuals specifically looked at area 24b of the supracallosal anterior cingulate cortex (ACC) and found a reduction in glia density (-22%) and neuronal size (-23%) in layer VI of the MDD subjects (Cotter et al., 2001). There were notable differences between the groups: in schizophrenia subjects, glia density (-20%) was reduced in layer VI before adjusting for multiple comparisons and there was no change in neuronal size, while BD subjects did not have a reduction in glia density or neuronal size. Neuronal density was similar in all the groups.
Cotter et al. (2002) examined postmortem dlPFC (BA 9) samples from MDD (n=15), BD (n=15), schizophrenia (n=15), and control (n=15) subjects. MDD was associated with reduced glia density (-30%) in layer V and reduced neuronal size (-20%) in layer VI. Similarly, glia density was reduced (-34%) in layer V of schizophrenia subjects and neuronal size was reduced in layers V (-14%) and VI (-18%) of BD subjects. At the time of this study, one of the hypotheses stemming from the neurotrophic theory of mood disorders was that the spatial arrangement of glia around neurons may be altered in depression since glial activity provides neurotrophic support to neurons. However, this study did not observe any change in the clustering of glia near neurons.
In postmortem samples from older-age (>60 years old) MDD subjects (n=15), there was a 30% reduction in OFC pyramidal neuron density in layers I-VI compared to control subjects (n=11) (Rajkowska et al., 2005). In particular, pyramidal neuron density was reduced in layers 3c and 5 in MDD subjects, whereas there was no impact on the density of nonpyramidal neurons and glia.
To assess the impact of depression on cortical GABAergic neurons, Rajkowska et al. (2006) examined calbindin-immunoreactive (CB-IR) and parvalbumin-immunoreactive (PV-IR) neurons in the dlPFC and OFC of postmortem samples from MDD subjects (n=14) and controls (n=11). CB-IR density was evaluated in layers 2 and 3a, while PV-IR density was evaluated in layers 3 and 4. Depressed subjects had reduced CB-IR density (-50%) in the dlPFC and reduced neuronal size, with a trend towards a reduction in the OFC, whereas there was no different in PV-IR density in the dlPFC nor in the size of PV-IR neuronal soma in the dlPFC or OFC.
2.3 Physiology of Depression
Early History
Some of the early, influential conceptions of depression originate with observations of what the earliest modern antidepressants did. The discovery of modern antidepressants and their effects was largely serendipitous, not part of a careful psychiatric medicine development program (Klein, 2008). Iproniazid, a drug used for tuberculosis, was unexpectedly found to improve mood in tuberculosis patients who had depressive symptoms (Hirschfeld, 2012); subsequently iproniazid was demonstrated to be a monoamine oxidase inhibitor (MAOI). And imipramine, a tricyclic antidepressant (TCA) known at the time as G 22,355, was originally made as a derivative of the antipsychotic chlorpromazine (Ban, 2006). Though it was structurally similar to chlorpromazine, it did not alleviate psychosis.
Before giving up on the drug, the Swiss psychiatrist Ronald Kuhn, who had originally encouraged the pharmaceutical company Geigy to test the drug in psychosis, tried the substance on a female patient with severe depression in 1956—it significantly alleviated her symptoms (Ban, 2006). The drug proceeded to work on dozens of patients and Kuhn consistently observed that their depression returned when imipramine was discontinued and subsided again when it was resumed. His first paper on the treatment of depression with imipramine was published in 1957. By the end of that same year it was released for clinical use in Switzerland with the brand name Tofranil.
In the span of a few years in the early 1950s, iproniazid’s MAOI properties were discovered and the presence of the endogenous monoamines serotonin and norepinephrine was demonstrated in the brain (Ban, 2006). The combination of those discoveries—that monoamines exist in the brain and that drugs which inhibit or alter their activity alleviate depression—contributed to the first major neuroscience-based understanding of depression. Unlike imipramine and iproniazid, the antihypertensive reserpine reportedly induced “severe depression” in some patients when high doses were given, an effect that was linked to its depletion of norepinephrine and serotonin (Schildkraut and Kety, 1967).
These reports made it easy to conceive of depression as an endogenously originating deficit in neurochemicals, especially serotonin and norepinephrine, along with dopamine to a lesser degree (Schildkraut and Kety, 1967). As the theory goes, if you have too little monoamine activity you will be depressed and if you reverse that decline depression will subside. Further, excessive norepinephrine activity was blamed for mania partly because reserpine reduced mania and “excitement,” placing depression and mania on a spectrum of mood states tied to neurotransmitter activity. It did not take long for holes in the hypothesis to appear.
For one, monoamine elevating drugs tended to have a therapeutic lag of at least 2-3 weeks (Hindmarch, 2001). If simply increasing neurotransmitters is all that is needed for efficacy, why would it take time for the effects to kick in? Revisions of the basic monoamine theory of depression and alternative hypotheses developed in the following decades. Many of the new ideas emphasized the detrimental effects of stress and how modulation of neuroplasticity may explain both depression and its alleviation (Hindmarch, 2001).
2.4 Neurotrophic Theory
One of the leading theories of depression is that stress and trauma—along with other behavioral, environmental, and genetics factors known to be associated with depression—result in altered neuroplasticity, neurogenesis, and neuronal survival. In some key regions like the PFC and hippocampus, neuroplasticity is reduced, impairing the ability to strengthen and weaken neuronal connections; in some areas, such as the nucleus accumbens (NAc), neuroplasticity may be upregulated. This is closely connected to changes in the activity of chemicals that affect neuronal survival, growth, and synaptic plasticity, known as neurotrophins. Among the key neurotrophins are brain-derived neurotrophic factor (BDNF), nerve-growth factor (NGF), and neurotrophin-3 (NT-3). A theory of depression based on neurotrophins and neuroplasticity took off in the 1990s (Duman et al., 1997).
Stress and its associated hormones, like the glucocorticoid cortisol, can initially support neuroplasticity and learning, yet with excessive and/or prolonged stress there is a reduction in BDNF and atrophy of stress-sensitive brain regions (Duman et al., 1997). Glucocorticoids are released from the adrenal glands, with stressors increasing their release. As they circulate the body, glucocorticoids bind to glucocorticoid and mineralocorticoid receptors; in the brain, those receptors are located at nuclear and plasma membrane sites (Popoli et al., 2011). When the effect of stress hormones is opposed, such as by performing an adrenalectomy (removal of the adrenal glands), BDNF expression increases (Duman et al., 2006).
Animal and human studies have repeatedly found depression is associated with reduced hippocampal and PFC size (Duman, 2012; Sheline et al., 1996; Rajkowska et al., 2005). Since both regions are important for cognition and mood, this deficit may underlie many of the key symptoms of depression or at least be a visible outcome of the same processes responsible for those symptoms.
Long-term treatment with standard antidepressants reverses stress- and depression-related neurological changes. For example, animals exposed to chronic antidepressant treatment exhibit an increase in the rate of neurogenesis in the hippocampus (D’Sa and Duman, 2002).
Human Research
Neurotrophins were reduced in postmortem hippocampus and PFC samples from suicide victims (n=30) compared to control subjects (n=24) (Karege et al., 2005). The suicide victims consisted of 10 depressed subjects who were negative for antidepressants at the time of death, 10 suicide victims with non-depression psychiatric disorders, and 10 depressed subjects who were positive for antidepressants. Levels of BDNF and NT-3 were reduced in the hippocampus of suicide victims who did not test positive for antidepressants, and BDNF was reduced in the PFC. In contrast, patients who tested positive for antidepressants did not differ from control subjects.
Kang et al. (2012) reported a reduction in the expression of genes related to synaptic function (CALM2, SYN1, RAB3A, RAB4B, and TUBB4) in postmortem dlPFC samples from MDD patients (n=14) compared to controls (n=14). The reduction among patients correlated with fewer synapses. Expression of the transcription factor GATA1 was increased in MDD and when expressed in rat PFC neurons, GATA1 decreased synapse-related gene expression, produced a reduction in dendrites and spines, and induced depressive behavior. This suggests the atrophy and impaired synaptic function associated with depression could be partly mediated by altered expression of synapse-related genes.
Animal Research
Frontal cortex BDNF mRNA expression was increased by acute electroconvulsive seizure (ECS) treatment in male rats (Niguya et al., 1995). Chronic ECS (10 days) reduced the induction of BDNF and tyrosine receptor kinase B (TrkB; receptor for BDNF) mRNA caused by acute ECS, but it also prolonged their expression for at least 18 h; in contrast, acute ECS by itself did not have an effect 18 h later. Chronic ECS amplified and prolonged the increase in BDNF mRNA in the hippocampal CA3 and CA1 pyramidal layers caused by acute ECS.
Similarly, chronic (21-day) treatment with the antidepressants tranylcypromine, sertraline, desipramine, or mianserin increased BDNF mRNA and every drug aside from mianserin increased TrkB mRNA in the hippocampus (Niguya et al., 1995). Non-antidepressants (cocaine, morphine, haloperidol) failed to replicate this effect. Chronic antidepressant and ECS also attenuated stress-induced downregulation of BDNF mRNA in the hippocampus.
BDNF deletion in the forebrain of mice blocked the antidepressant effect of desipramine in the FST—three injections were given, specifically at 24, 4, and 1 h before testing—and interfered with hippocampus-dependent learning and long-term potentiation (LTP), an aspect of synaptic plasticity (Monteggia et al., 2004).
Instead of neurogenesis driving antidepressant effects, Bessa et al. (2009) identified an apparent role for altered neuron structure and synaptic plasticity in the PFC and hippocampus. CMS exposure caused depressive behavior and impaired neurogenesis in rats; both changes were improved by 1-2 weeks of treatment with antidepressants like imipramine and fluoxetine. The number of recently mitotic neurons (Ki67+ and BrdU+) in the hippocampus was reduced by CMS, whereas antidepressants reversed the reduction and increased neurogenesis above baseline.
However, impeding the neurogenic effect of antidepressants with methylazoxymethanol did not block their behavioral effects (Bessa et al., 2009). Stress-induced depressive behavior correlated with a loss of synaptic contacts and dendritic atrophy, not just reduced neurogenesis. Antidepressants attenuated dendritic atrophy in regions of the PFC and hippocampus, and they attenuated a reduction in the proportion of larger ‘mushroom’ dendritic spines relative to thin spines.
2.5 The Role of Glutamate
Studies in the 1990s found NMDAR antagonists caused rapid antidepressant-like effects in animals (Trullas and Skolnick, 1990). For example, the effect of imipramine on depression-related behavior in mice was mimicked by the competitive NMDAR antagonist AP-7 and the noncompetitive antagonist MK-801. Importantly, chronic but not acute exposure to 17 classic antidepressants (including imipramine, citalopram, and ECT) altered NMDAR activity in mice, most prominently in the cerebral cortex (Skolnick et al., 1996). Those NMDAR adaptations developed slowly with repeated antidepressant administration and persisted for some time after treatment was discontinued.
Other researchers observed the same. Nowak et al (1993) showed chronic imipramine reduced NMDAR ligand binding in the cerebral cortex of mice, but not in the hippocampus, striatum, or basal forebrain (which includes the NAc). Similar results were reported in rats, where chronic imipramine reduced NMDAR ligand binding, as did ECT, yet acute treatment with either was ineffective (Paul et al., 1993).
The observation that NMDAR alteration was one of the adaptations caused by diverse antidepressants and that NMDAR antagonists themselves had preclinical signs of therapeutic potential led some researchers to hypothesize NMDAR downregulation is an important mechanistic pathway shared by many antidepressants. The glutamate/NMDAR hypothesis was bolstered by studies reporting glutamate receptor abnormalities in people with depression and in suicide victims (Nowak et al., 1995)
In the early 2000s there was interest in the NMDAR glycine site because it had been discovered that the NMDAR coagonists glycine and D-serine reduce the negative and cognitive symptoms of psychotic disorders when added to normal antipsychotics (Javitt, 2004). NMDARs had also been on the minds of researchers for quite some time in the context of psychosis because the effect of antagonists like ketamine and PCP was considered a model of psychotic disorders, potentially offering a way to research the physiology of those disorders. As such, schizophrenia was initially a major target among people interested in using glutamatergic drugs in psychiatry, although by the 2000s there was also a growing interest in the use of NMDAR antagonists for mood disorders.
2.6 Treatment Options (And Their Shortcomings)
History of Antidepressants and Psychiatric Medications
Drugs have been used for mood disorders, anxiety, and other psychiatric conditions for millennia, but the modern history of psychiatric drugs dates back to the 1800s and it was not until the mid-1900s that a more formal, evidence-guided field of psychiatric medicine developed.
Before the introduction of MAOIs and TCAs, seizure and shock therapies were very popular. In the 1930s, camphor was administered to induce seizures in psychotic patients, later being replaced by the circulatory stimulant pentylenetetrazol. Seizure-based therapies and insulin-induced coma were widespread by 1937, and the tolerability and therapeutic potential of convulsive therapies improved further after the introduction of electroconvulsive therapy (ECT) by the neurologist Ugo Cerletti and the psychiatrist Lucio Bini; ECT was shown to be more effective for mood disorders than for psychosis (Hirschfeld, 2012). The improved safety profile of ECT relative to drug-based convulsive therapies facilitated its adoption and it remains in use today.
At the same time as seizure therapies were spreading, amphetamine, which had originally been developed as a decongestant, was approved by the American Medical Association (AMA) for depression (Hirschfeld, 2012). It was undoubtedly effective and had a notable effect on anhedonia, but its short-lasting benefits, abusability, and side effects led to it falling out of favor by the 1960s, at which point other antidepressants were available.
The 1950s to 1960s saw the adoption of drugs like lithium, iproniazid, chlorpromazine, chlordiazepoxide (the first benzodiazepine), and the antipsychotic clozapine.
The Inadequacy of Current Treatments
Existing antidepressants are highly inadequate. Although they reduce symptoms for many people, a large minority of patients have treatment-resistant depression (TRD), meaning they have not adequately responded to at least a couple antidepressants. Many TRD patients also do not respond to augmentation therapies (e.g. antipsychotics or lithium) or ECT. For depressed patients who do respond to common treatments like selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), those drugs still have shortcomings, particularly 1) a lag of weeks to months before the benefits are fully realized and 2) comparatively poor efficacy in certain symptoms, such as anhedonia.
The lag can be highly consequential if someone is suicidal—some antidepressants may even exacerbate depression and anxiety symptoms early in treatment, increasing suicide risk for some patients (Jick et al., 2004)—and it reduces treatment adherence because people become frustrated by the inefficacy and side effects, resulting in premature discontinuation of therapy and reluctance to try other medications.
In one of the largest trials of antidepressants, the open-label STAR*D trial of 2,876 patients who were treated with flexible-dose citalopram (an SSRI) for 14 weeks, the overall remission rate was only 28% and the response rate was 47% (Trivedi et al., 2006). In depression studies like this one, remission usually means someone no longer meets the criteria for depression, while response means their symptoms have been reduced by ≥50%. For those who responded to citalopram, the mean time to response was ~6 weeks. Responders then exhibited a 40% relapse rate over the next 12 months of naturalistic follow-up, with a mean time to relapse of 4.1 months (Rush et al., 2006).
The STAR*D trial was useful because it included people with comorbid psychiatric issues like substance use and anxiety disorders, whereas many clinical studies do not permit those comorbidities, thereby making their results less generalizable to real world settings. STAR*D also provided insight into treatment-resistance because those who did not remit with citalopram were able to receive up to three more levels of therapy. The other treatments included bupropion, venlafaxine, sertraline, nortriptyline, tranylcypromine, and cognitive therapy, as well as augmentation options like lithium, T3 thyroid hormone, and buspirone.
With four levels of treatment, the cumulative theoretical remission rate was 67% (Gaynes et al., 2009), and that rate was inflated due to a high dropout rate and methodological flaws (Pigott, 2015). Even with the very charitable remission rate of 67%, that means 33% of people with depression will not remit with up to four standard treatments and more than half will either not experience remission or will not maintain their improvement for a year (Rush et al., 2006). Therefore, going by the STAR*D trial, only a minority of people experience sustained remission from common antidepressants with up to four treatments.
“Remission” also does not mean an absence of depressive symptoms or persistent side effects from treatment. 90% of patients who remitted during the first level of treatment with citalopram still had at least one residual symptom, with a median of three (Nierenberg et al., 2009). The most common symptoms were weight increase and middle-of-the-night insomnia (aka middle insomnia). Many of the subjects reporting middle insomnia did not initially have it as one of their baseline depression symptoms either; 25% of those who did not have middle insomnia at the start of the study developed it by the end. Other seemingly treatment-emergent symptoms included appetite change, decreased concentration or interest, decreased energy, and hypersomnia.
In a separate analysis of more than 100 controlled trials (average duration of 7.2 weeks) encompassing 17,000 antidepressant-treated and 10,000 placebo-treated unipolar depression patients, the response rate with common FDA-approved antidepressants averaged 54%, compared with 37% with placebo (Undurraga and Baldessarini, 2012), suggesting ~17% of people experiencing a major depressive episode who would not respond to placebo will respond to an antidepressant in the short-term.
Based on studies like these, the burden of untreated and poorly treated depression is high. This is likely the result of it being difficult to fix depression with drugs combined with a lack of innovation in psychiatric medicine. The modern era of pharmacotherapy for depression began 70 years ago, yet the most commonly prescribed drugs at present do not offer superior efficacy to those that were available in the 1950s and 1960s. Standard treatments like SSRIs and SNRIs are in many ways safer than the early TCAs and MAOIs, but they are only similarly effective or somewhat less effective, leaving many people in need of new options.
ECT
It is estimated that more than 100,000 patients in the US receive ECT each year and studies suggest it may be fairly effective in TRD, producing a response in >60% of patients (Taylor, 2008). In a 6-week randomized controlled trial (RCT) of paroxetine (n=18) compared with ECT (n=21) for TRD, there was a much larger response rate with ECT (71% vs. 29%) (Folkerts et al., 1997). ECT was typically administered six times over a 2-week period.
ECT has been controversial for decades, in large part due to cognitive impairment that many people claim is the result of brain damage. A meta-analysis of 84 studies (2981 patients) identified impairments in episodic memory and executive function, particularly in the first three days after ECT, but within two weeks those impairments resolved and some areas of cognitive function appeared to improve (Semkovska and McLoughlin, 2010). However, there is still far too little research on the long-term effects of ECT, including post-acute efficacy and hypothetical risks like persistent amnesia, which unfortunately can be said for most psychiatric treatments.
Section 3: Introduction to Ketamine
3.1 Pharmacodynamics
NMDAR
The most well-known mechanism of ketamine is NMDAR antagonism. Ketamine exists as a racemate, meaning it has two isomers (specifically, enantiomers) that function as two distinct drugs. Those isomers are S-ketamine and R-ketamine, with the former being a stronger NMDAR antagonist than the latter. The NMDAR affinity (Ki) of S-ketamine (0.30 μM) is around 4x greater than R-ketamine (1.4 μM) (Ebert et al., 1997). Because there are different NMDAR subtypes with different subunit compositions, ketamine has somewhat different affinities depending on the subtype. There is also a large effect of magnesium levels such that the affinity and subtype preference of ketamine changes when factoring in the concentration of magnesium in the body (Kotermanski and Johnson, 2009).
In the presence of physiological levels of magnesium (1 mM), ketamine has a fair degree of subunit selectivity and its affinity for GluN2A- and GluN2B-containing NMDARs is reduced significantly more than its affinity for GluN2C- and GluN2D-containing receptors, which is caused by magnesium producing variable levels of inhibition depending on the receptor subtype (Kotermanski and Johnson, 2009). The IC50 (μM) values without and with magnesium are as follows: GluN2A (0.33 vs. 5.35), GluN2B (0.31 vs. 5.08), GluN2C (0.51 vs. 1.18), GluN2D (0.83 vs. 2.95).
This suggests ketamine is relatively selective for GluN2C-containing NMDARs and those receptors could be particularly relevant to its mechanism of action. Khlestova et al. (2016) estimated the dissociative and psychotomimetic effects of ketamine occur when the extracellular concentration in the brain is ~0.5 μM, which would inhibit GluN2C-NMDARs by ~30%, while only inhibiting GluN2A- or GluN2B-NMDARs by ~10%. That estimate assumes the serum and brain concentrations are similar, but there are studies demonstrating a brain:serum ratio of >2 and even 6.5:1 (Cohen et al., 1973), bringing the relevant concentration to upwards of 1.0 μM. Regardless, at any of the estimated brain concentrations, ketamine would be expected to impact GluN2C-NMDARs more than GluN2A- and GluN2B-containing receptors. The major caveat to this is that the expression of NMDAR subtypes in the brain is biased towards GluN2B and GluN2A receptors, attenuating the impact of its selectivity.
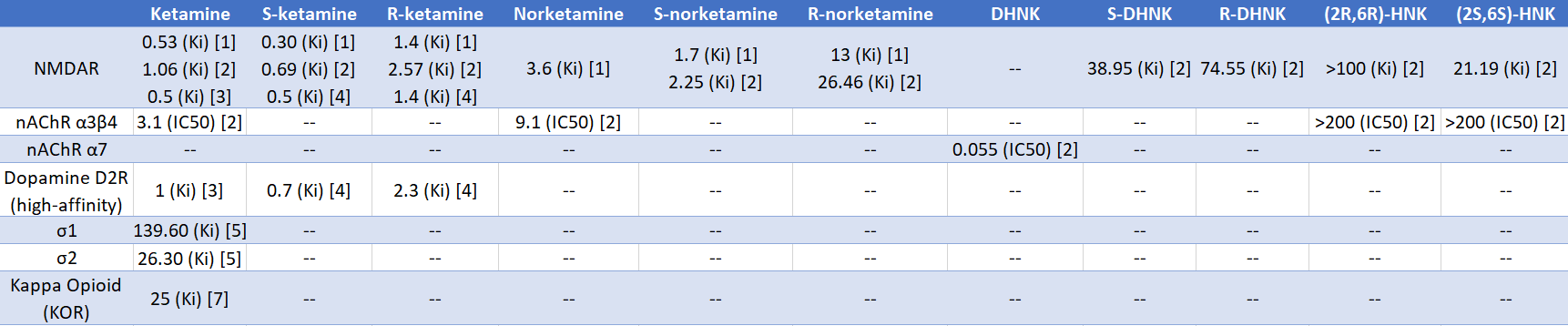
Ketamine is not selective for NMDARs, but there are few other targets reliably affected by the drug, as shown by Salat et al. (2015). Out of 80 targets, ketamine and norketamine at 10 μM primarily affected NMDARs, with ketamine displacing 88% of PCP binding and norketamine displacing 56% of binding. Ketamine did not affect muscarinic receptors, opioid receptors (KOR, DOR, MOR), sigma receptors (σ), serotonin receptors, or monoamine transporters (SERT, NET, DAT). Using MK-801, an NMDAR binding assay found the affinity (Ki) of ketamine was 0.119 μM, while it was 0.97 μM for norketamine and 3.21 μM for DHNK.
Acetylcholine Receptors
Ketamine (0.1-1 μM) does not inhibit acetylcholine-induced currents mediated by rat α7 nicotinic acetylcholine receptors (nAChRs), but some of its metabolites have an appreciable effect (Moaddel et al., 2013). α7 nAChR currents were reduced by 60% with 100 nM of DHNK and by 45% with norketamine; the IC50 of DHNK was 55 nM. The IC50 for suppression of S-nicotine induced currents mediated by rat α3β4 nAChRs was 3.1 μM for ketamine and 9.1 μM for norketamine, whereas DHNK, (2S,6S)-HNK, and (2R,6R)-HNK were inactive, indicating their IC50 are >200 μM.
Binding Data
See Table 1.
3.2 Pharmacokinetics
Overview
Bioavailability
- Intramuscular (IM) = 93% (Clements et al., 1982)
- Oral = 17% (Clements et al., 1982); 24% (Chong et al., 2009); 20% (Yanagihara et al., 2003)
- Sublingual = 24% (Chong et al., 2009); 30% (Yanagihara et al., 2003)
- Intranasal = 45% (Yanagihara et al., 2003)
- Rectal = 30% (Yanagihara et al., 2003)
Elimination Half-Life
- IV = 120 min (Yanagihara et al., 2003)
Kinetics and Metabolism
Ketamine is extensively metabolized via the action of CYP450 enzymes in the liver. N-demethylation yields norketamine, which is subsequently metabolized to dehydronorketamine (DHNK) and a variety of hydroxynorketamines (HNKs) (Zanos et al., 2018).
Human Research
After the infusion of an antidepressant dose of ketamine (0.5 mg/kg IV, 40-min), ketamine, norketamine, DHNK, and (2R,6R;2S,6S)-HNK are present in plasma during the first 230 min and significant levels (>5 ng/mL) remain at 24 h, whereas plasma (2S,6S;2R,6R)-hydroxyketamine and (2S,6R;2R,6S)-hydroxyketamine were unquantifiable, based on a study of nine BD patients (Zhao et al., 2012). There was substantial interpersonal variation for metabolite levels and half-lives. At 230 min, the major metabolite was DHNK in four, norketamine in three, and (2S,6S;2R,6R)-HNK in two. Consistent with norketamine being metabolized to DHNK and to HNKs, the concentration of norketamine declined from the end of the infusion to the 230 min timepoint, whereas DHNK and (2S,6S;2R,6R)-HNK increased.
Norketamine exposure is much higher with oral administration compared to IV or intranasal. Yanagihara et al. (2003) studied three healthy males who were given 20 mg IV, 50 mg oral, 50 mg sublingual, 50 mg rectal, and 25 mg intranasal; the intranasal route involved an aqueous solution delivered through an atomizer. The area under the curve (AUC), i.e. total exposure integrated over time, for norketamine was 500 ng*h/mL with the oral route, compared with 280-460 ng*h/mL for rectal and sublingual, and only 100 ng*h/mL with intranasal. The ketamine enantiomers were detected in plasma within 5-20 min with oral, sublingual, rectal, and intranasal use, and they were below the detection limit at 8 h; norketamine was present by 10-30 min via those routes. Unlike in other studies, there were no significant kinetic differences between the ketamine and norketamine enantiomers, which could be related to the sample size or to genetic differences since this study used Japanese volunteers.
A study of 10 healthy males who were administered R-ketamine (7 mg IV, 30-min) and S-ketamine on separate days found their kinetics were similar, but clearance was 20% lower for R-ketamine (Persson et al., 2002). Six subjects had a phenotype of extensive CYP2D6 and CYP2C19 metabolism, while there were two CYP2D6 poor metabolizers and two CYP2C19 poor metabolizers; metabolic status did not affect kinetics. At the end of the infusion, the peak arterial concentration (Cmax) was 72 ng/mL for S-ketamine and 81 ng/mL for R-ketamine, compared to venous values of 51 ng/mL for S-ketamine and 58 ng/mL for R-ketamine. Initially the arterial concentration was 50% higher than the venous level, but it also declined faster and fell below the venous concentration 10 min post-infusion.
With a 5-day continuous infusion of ketamine (40 mg/h) for the treatment of complex regional pain syndrome (CRPS), the plasma concentrations at Day 3 were (in ng/mL): 12,496 for S-ketamine, 16,214 for R-ketamine, 27 for S-norketamine, 38 for R-norketamine, 109 for S-DHNK, 153 for R-DHNK, 656 for (2S,6S;2R,6R)-HNK, and 881 for (2S,6R;2R,6S)-HNK (Moaddel et al., 2010).
Animal Research
The brain Cmax of ketamine (20 mg/kg IV) is reached within one minute of IV administration in male rats and brain accumulation occurs, with a brain:plasma ratio of 6.5:1 (Cohen et al., 1973). Norketamine also accumulates in the brain, with the brain concentration surpassing plasma 10 min after injection. Brain distribution was studied using 20 mg/kg (IV) because 60 mg/kg was near the LD50 in these animals. Ketamine’s brain levels started to fall shortly after administration, but the 6.5:1 brain-to-plasma ratio was maintained for at least 10 min. More ketamine was detected in the cerebral cortex compared to the midbrain, brainstem, and cerebellum at early timepoints (30 sec, 1 min), becoming equally distributed afterwards.
Norketamine was first detected in plasma at 30 sec and it reached a plateau of 2.25 μg/mL between 5-10 min. In the brain it was first detected at 1 min and it increased in concentration during the 10-min experiment, with a brain:plasma ratio of 2.5:1 at 10 min (Cohen et al., 1973). Brain tissue could not metabolize ketamine, but liver homogenates metabolized it to norketamine.
Within 10 min of ketamine (10 mg/kg IP) administration in male mice, there were quantifiable plasma concentrations of ketamine, norketamine, (2S,6S;2R,6R)-HNK, (2S,6R;2R,6S)-HNK, and DHNK (Can et al., 2016). The kinetic profiles of ketamine and its metabolites in plasma and the brain were as follows:
Table 2. Plasma kinetics of ketamine and its metabolites after 10 mg/kg (IP) in male mice (Can et al., 2016).
Table 3. Brain kinetics of ketamine and its metabolites after 10 mg/kg (IP) in male mice (Can et al., 2016).
Plasma levels of other HNK metabolites were below the limit of quantitation at all timepoints, including (2S,5S;2R,5R)-HNK, (2S,4S;2R,4R)-HNK, (2S,4R;2R,4S)-HNK, and (2S,5R;2R,5S)-HNK (Can et al., 2016). The brain Cmax of ketamine was 73% larger than the plasma Cmax, whereas the brain Cmax was 49% smaller for norketamine and 45% smaller for (2S,6S;2R,6R)-HNK. DHNK was below the limit of quantitation in the brain.
3.3 Medical Effects of Ketamine
Anesthesia
For decades, ketamine has been a popular anesthetic in humans and in veterinary settings. Most often it is delivered intravenously, but it can be given intramuscularly, albeit with some injection site pain, a longer recovery time, and a greater likelihood of vomiting (Marland et al., 2013). Anesthesia usually begins with 1-2 mg/kg (IV) or 8-10 mg/kg (IM). Its dissociative anesthetic effects have an IV onset of 1-2 minutes. Ketamine is used in prehospital and hospital settings, as well as in low-income and emergency settings, such as during disaster relief when working with limited equipment and in combat situations.
It is similarly effective to opioids for analgesia without impairing respiration and its cardiovascular effects are typically limited to a modest increase in blood pressure (BP) and heart rate (HR), which is not a concern for most patient populations. Part of its popularity stems from it being relatively easy to work with because of its respiratory and cardiovascular stability. Emergence phenomena (e.g. agitation, dissociative symptoms) occur in 1-2% of children and 10-20% of adult patients (Marland et al., 2013). Other adverse effects include muscle hypertonicity and vomiting (5-15% of patients).
Some of ketamine’s adverse effects can be reduced by other medications. The antiemetic ondansetron reduces vomiting and propofol can counteract its cardiovascular stimulation and attenuate some of the emergence phenomena. The combination of ketamine and propofol is fairly common and they are used together under the name Ketofol.
Analgesia
Prevention of Chronic Pain
Ketamine may be a good analgesic in some cases of neuropathic pain, chronic pain, and when given around the time of an injury or operation. There is a hypothetical basis for using ketamine to prevent the development of chronic pain after an injury or surgery. Chronic pain is thought to involve a ‘wind-up’ mechanism in which nociceptive (pain-processing) pathways along the spinal cord become sensitized, causing long-term amplification of pain even in the absence of a persistent injury. NMDAR activation contributes to wind-up in rat dorsal horn (a region of the spinal cord) neurons; this was inhibited by the NMDAR antagonist kynurenate (Davies and Lodge, 1987). Therefore, the NMDAR antagonism of ketamine could hypothetically inhibit this process in humans.
Using ketamine around the time of an operation to prevent chronic pain currently lacks strong supporting evidence in humans. Epidural ketamine with bupivacaine was not superior to bupivacaine alone for reducing persistent postoperative pain after lower limb amputation (Wilson et al., 2008). It affected pain sensitivity eight days after the operation, but not at later timepoints up to one year. A trial comparing S-ketamine infusion (0.1 mg/kg/h IV; 60 h) with placebo after thoracotomy (surgery to open the chest wall) found no difference in persistent pain at 1, 3, or 6 months, but S-ketamine was associated with a reduced need for supplementary analgesia on the day of the operation and on the subsequent day (Mendola et al., 2012). Similarly, Duale et al. (2009) studied the effects of a 24 h ketamine infusion in 86 patients undergoing thoracotomy and found it was only superior to placebo during the first 24 h after the operation, with no significant effect on analgesic use or pain six weeks and four months after the surgery.
Reducing Postoperative Pain and Opioid Use
A reduction in acute postoperative pain and a concomitant reduction in the need for standard analgesics (e.g. opioids) are more consistently supported effects of ketamine. In a Cochrane review of 130 studies encompassing 4600 people given perioperative ketamine compared with 3800 control patients, ketamine was associated with a 19% reduction in postoperative opioid use at 24 h and 48 h, a 14-22% reduction in pain at rest and during movement at 24 h and 48 h, and an increase in how long it took for patients to first request a postoperative analgesic (Brinck et al., 2018). Ketamine use also correlated with a small reduction in the incidence of postoperative nausea and vomiting (23% with ketamine vs. 27% with placebo).
Chronic Pain
Ketamine has shown variable effects in chronic pain, partly dependent on the underlying condition. In fibromyalgia patients, S-ketamine (0.5 mg/kg IV, 30-min) only produced pain relief for slightly longer than the duration of the infusion, but in studies of CRPS, long infusions have sometimes produced persistent benefit. For example, pain was reduced for months in CRPS patients treated with either a 100-hour infusion of S-ketamine (up to 20-30 mg/h) or with a 4-hour daily infusion for 10 days (Niesters et al., 2014).
3.4 General Effects of Ketamine
Healthy People
Many studies have examined the effects of subanesthetic and anesthetic doses of ketamine in healthy people. It produces a short-lasting altered state that can include hallucinations, dissociation, and sometimes psychotomimetic effects, particularly paranoia, ideas of reference, and other unusual patterns of thinking. The experience can be pleasant or disturbing, but in either case the greatest effects rarely persists much beyond 1-2 hours. There are few to no cases in which acute ketamine exposure in a psychiatrically healthy person produced or triggered a long-lasting disorder, although a small minority of people may make poor and potentially dangerous choices under the influence.
Its effects on mood are variable and may significantly depend on the user, the environment, and the dose. Studies often report transient mood elevation, but others have found ketamine can induce a dysphoric mood and be disliked more than placebo (Lofwall et al., 2006; Krystal et al., 2005). Because mood elevation is a common effect in recreational settings and ketamine would likely be much less popular if it consistently caused dysphoria, these results could be attributable to people receiving ketamine in a non-recreational research setting that is not particularly conducive to enjoyable experiences.
Krystal et al. (1994) performed a double-blind, placebo-controlled (DBRCT) crossover trial of ketamine (0.1 and 0.5 mg/kg IV, 40-min) in 19 healthy people. Primarily at the 0.5 mg/kg dosage, ketamine produced positive and negative psychotic-like symptoms, altered perception, dissociation, and impaired cognitive performance on tests of vigilance, verbal fluency, abstract reasoning, and word recall after a 10 minute delay—although it did not impair immediate recall and recall after distraction. Before the perceptual and cognitive changes were fully developed, some subjects described ketamine as feeling similar to alcohol.
Psychotic thinking patterns were present in 7/19 participants (Krystal et al., 1994). They experienced ideas of reference and frequently were paranoid about staff members, believing they had malevolent intentions. Subjects experienced a state of amotivation and they struggled to start or sustain interpersonal interactions like conversations; often they would emotionally withdraw from others and they appeared to be experiencing dulled emotions.
Ketamine’s effect on perception mostly involved illusions, either of the self or the environment (Krystal et al., 1994). During the experience, it was common for people to experience depersonalization, derealization, and a sense of losing control of their thinking; they also perceived their body parts or body shape as altered and their environment looked unusual: colors changed and objects appeared distant. The psychotomimetic, dissociative, and hallucinatory effects substantially declined or resolved within 15 min after the infusion. No subject had persistent physiological or psychological problems after ketamine exposure. The acute hemodynamic effects were not an issue. Ketamine caused relatively minor dose-related increases in systolic and diastolic blood pressure, and heart rate did not significantly increase. Blurred vision was reported by 44% of participants on the high-dose day and half of those participants also had horizontal nystagmus.
Likewise, other studies of subanesthetic ketamine in healthy people reported transient impairments in cognition (e.g. memory and attention), impaired balance, and acute psychotomimetic and hallucinogenic effects (Malhotra et al., 1996; Lofwall et al., 2006). Molhatra et al. (1996) found the psychotomimetic effects were most notable during the first hour of a 1 h IV infusion (0.77 mg/kg/h)—the effects significantly decreased within 30 min post-infusion.
Lofwall et al. (2006) used an intramuscular administration method instead of IV. In a DBRCT of ketamine (0.2 and 0.4 mg/kg IM) versus placebo in 18 people, ketamine did not cause frank hallucinations or mystical experiences, but it did alter cognition and perception, with larger effects at 0.4 mg/kg. The hallucinatory and psychotomimetic effects were limited to distortions of the environment and a sense of dissociation instead of frank hallucinations and delusions (i.e. fixed, false beliefs), making it somewhat distinct from schizophrenia and other psychotic disorders that involve a larger burden of full hallucinations and delusions. Interestingly, despite being told they may have mystical experiences, no participant experienced them. The absence of mystical experiences could have been partly attributable to the research setting.
In a large observational study, Xu et al. (2015) compared the effects of ketamine in those who had taken it acutely (n=135) or chronically (n=187) to the natural symptoms of early schizophrenia patients (n=154) and chronic schizophrenia inpatients (n=522). The chronic ketamine users had been admitted due to ketamine dependence; they had an average daily intake of 3.4 grams, 75% were daily users, and 86% used other substances (e.g. MDMA, amphetamine). In contrast, the acute ketamine group was otherwise healthy and received a single dose in a randomized manner. The early schizophrenia group consisted of people who had been psychiatrically hospitalized for the first time and did not have a history of medication use, whereas the chronic patients were all being treated with antipsychotics.
Similar positive, negative, cognitive, and dissociative symptoms were seen across the groups, with greater similarly between chronic ketamine users and schizophrenia patients (Xu et al., 2015). Symptoms were much milder overall in ketamine users compared to patients, and psychotic symptoms were greater in the chronic ketamine group versus the acute ketamine group. Among individual symptoms, chronic ketamine users had a particular increase in ratings on emotional items (e.g. anxiety, feelings of guilt, and depression) and somatic items.
People with Psychotic Disorders
Because ketamine produces psychotomimetic effects and has been used as a model for psychotic disorders, it is conceivable the drug could produce uniquely strong effects in people with underlying psychotic disorders and perhaps aggravate their illness even after the acute drug effects wear off. Life stressors are known to be an aggravating factor in psychosis, serving as triggers that activate or exacerbate symptoms—it makes sense that ketamine could also be a trigger. However, most of the research on this topic shows ketamine rarely produces lasting negative effects in people with schizophrenia. It acutely increases their symptoms and can reactivate positive symptoms from earlier in their illness or before medication use, but those reactivations tend to dissipate within hours or days.
In a DBRCT of nine schizophrenic patients that compared ketamine (0.1, 0.3, and 0.5 mg/kg IV) with placebo, ketamine caused a transient dose-related increase in psychotic symptoms, with an especially large increase in positive symptoms (e.g. hallucinations, delusions, thought disorder) at 0.3 and 0.5 mg/kg rather than negative symptoms (Lahti et al., 1995). The effects were similar to what they had experienced during earlier episodes of their natural illness. Although these patients were initially studied while on haloperidol, six were retested after being off medication and they had similar effects, with no evidence of a dampening influence from haloperidol. Follow-up interviews at 8 and 24 h showed 4/9 patients had a spontaneous recurrence of psychotic effects after ketamine wore off, but the effects were less intense than during the acute effect period.
Malhotra and colleagues (1997) studied the effects of ketamine (0.12 mg/kg IV bolus, then 0.65 mg/kg over 1-hour) in antipsychotic-free schizophrenic patients (n=13) and healthy controls (n=16). There were transient increases in positive and negative psychotic symptoms, along with altered bodily perception and/or time perception and thought disorganization; the effects were stronger and more psychosis-like in the patients. Schizophrenic subjects experienced auditory hallucinations and paranoia, unlike control subjects; auditory hallucinations occurred in 8/13 patients, including four patients who had not been actively hallucinating before the study. The patient group also had larger deficits in free recall and recognition memory than the control group.
Ketamine largely reactivated or amplified symptoms that the patients had previously experienced in their illness instead of producing novel symptoms (Malhotra et al., 1997). Importantly, ketamine did not cause auditory hallucinations in the five schizophrenic patients who did not have a history of that symptom.
Preadministration/Blocking Studies
The mechanisms responsible for ketamine’s effects have been researched using preadministration/blocking studies in which another drug, such as an antipsychotic or a glutamate reducer like lamotrigine, is given alongside ketamine. If a drug inhibits ketamine’s effect, that could provide some insight into how ketamine is working.
In a DBRCT crossover study of ketamine (0.12 mg/kg IV bolus, then 0.65 mg/kg over 1 h) in schizophrenic patients (n=10) with and without clozapine treatment, medication-free patients had an increase in positive and negative psychotic symptoms (Malhotra et al., 1997). Those changes were greatly reduced by clozapine, with a particularly large reduction in thought disturbance. On specific ratings of thought disturbance, clozapine reduced conceptual disorganization, but not hallucinations or unusual thought content.
Krystal et al. (1999) examined the effects of haloperidol preadministration in healthy participants (n=20) who were given ketamine (0.26 mg/kg IV bolus, then 0.65 mg/kg for 1 h). That dose of haloperidol occupied ~70% of dopamine D2 receptors. Haloperidol reduced ketamine’s executive function impairment, but it failed to attenuate the psychotomimetic, euphoric, and perception altering effects. This is consistent with the observation by Lahti et al. (1995) that haloperidol did not significantly change ketamine’s acute effects in schizophrenic patients.
The mGluR2/3 agonist eglumegad (LY354740), which should reduce glutamate release by targeting glutamatergic autoreceptors, attenuated working memory impairment induced by ketamine (0.26 mg/kg IV bolus, then 0.65 mg/kg/h for 100-min) in 19 healthy people (Krystal et al., 2005). Eglumegad also attenuated ketamine-induced vigilance reduction and increased distractibility. However, most of ketamine’s effects were not changed by eglumegad. It did not affect impairments in immediate and delayed recall nor did it block ketamine-induced psychotomimetic effects.
In a DBRCT of ketamine (0.26 mg/kg IV bolus, then 0.65 mg/kg/h for 90-min) given to healthy subjects (n=16), there was a significant reduction in ketamine-induced dissociative (CADSS) effects, psychotomimetic (BPRS positive and negative) effects, and learning and memory impairment when lamotrigine (300 mg) was preadministered (Anand et al., 2000). In contrast, lamotrigine increased the mood-elevating effect of ketamine right after administration, but that interaction disappeared by the 30 min timepoint.
Imaging Studies
The acute psychotomimetic effects of ketamine in healthy people (n=17) correlated with a bilateral increase in PFC metabolic activity in a placebo-controlled FDG-PET study using a 0.12 mg/kg IV bolus of ketamine followed by a 1-hour infusion of 0.65 mg/kg (Breier et al., 1997). Aside from a small area of reduced activity in the right cerebellum, ketamine did not affect metabolic activity in other regions. A correlation between elevated prefrontal activity and psychotomimetic effects was specifically seen with ketamine’s impact on conceptual disorganization.
Elevated prefrontal activity was also observed in a study of healthy people (n=10) undergoing FDG-PET analysis with an initial IV infusion of 20 mg (5-min) followed by a continuous infusion of 0.02-0.03 mg/kg/min to maintain its psychotomimetic effects for one hour (Vollenweider et al., 1997). Metabolic activity increased in the frontomedial region and the ACC, with those changes positively correlating with psychotomimetic symptoms, particularly ego pathology. Whole brain glucose metabolism was increased by 25%, with variable effects depending on the region: the largest increases were in the ACC (+34%) and frontal cortex (30-34%), with smaller increases in the insula (+19-23%), parietal (+18-25%), somatosensory (+19-25%), motor (+15-18%), temporal (+16-19%), and occipitomedial (+16-17% cortices.
Ketamine’s hyperfrontal effect was confirmed by examining ratios of metabolic rates between regions (Vollenweider et al., 1997). There was an increase in frontomedial/occipitomedial (9-14%) and frontomedial/temporomedial (8-12%) ratios in both hemispheres. Among the psychotomimetic effects were hallucinations, thought disorder, apathy, derealization, and depersonalization; visual changes ranged from distortions to elementary and complex hallucinations. Most subjects became emotionally withdrawn during the infusion, had a flattened affect, and became concrete in proverb interpretation.
In a DBRCT crossover study of 10 healthy males, 1H-MRS was used to study ketamine’s effect on the bilateral ACC (Rowland et al., 2005). ACC glutamine increased during ketamine infusion (0.27 mg/kg IV 10-min, then 0.00225 mg/kg/min for up to 2 h), indicative of an increase in glutamate release; the increase did not correlate with ketamine’s positive or negative psychotomimetic effects. It increased dissociation (CADSS) and psychotomimetic (CADSS) scores, as well as ratings on the Scale for the Assessment of Negative Symptoms (SANS), but none of those effects correlated with the change in glutamine.
Using fMRI, Deakin et al. (2008) investigated the role of elevated glutamate activity as well as the connection between ketamine’s subjective effects and neural changes in a DBRCT of 33 healthy males. Ketamine was delivered as a 0.26 mg/kg IV bolus followed by an infusion of 0.25 mg/kg/h. Behaviorally, ketamine increased BPRS and CADSS scores, but the two were not highly correlated; the BPRS subscales for hallucinations and euphoria were elevated. Although noises were misperceived and normal actions were misinterpreted as suspicious, it did not produce frank psychosis.
Ketamine increased blood oxygen level dependent (BOLD) signal, an indicator of activity, in the precuneus (BA7), posterior cingulate gyrus (BA24), motor cortex (BA6), superior frontal gyrus (BA8), inferior temporal gyrus (BA20), hippocampus, and superior temporal gyrus (BA22) bilaterally, whereas a reduction occurred in the bilateral medial OFC (BA11) and temporal pole (BA38) (Deakin et al., 2008). The decrease in the medial OFC and temporal pole occurred within two minutes, with deactivation then spreading to the subgenual cingulate and anterior PFC (BA10), remaining reduced for seven minutes. By the third minute, there was significant activation of the anterior thalamus and multiple cingulate cortex regions (BA30, BA31, and BA23), extending to the posterior parahippocampal gyrus.
The dissociative effects (CADSS) correlated with the areas of deactivation, namely the OFC and subgenual cingulate; also, deactivation of the OFC correlated with the increase in psychotomimetic (BPRS) effects (Deakin et al., 2008). Among the areas of activation, the increase in frontal pole (left BA10) activity correlated most strongly with psychotomimetic ratings, and there were weaker correlations with the parahippocampal gyrus and posterior cingulate. Altered thalamic activity did not correlate with subjective effects.
Lamotrigine (300 mg oral) pretreatment reduced ketamine’s elevation in total BPRS score as well as the increase in BPRS thought disorder, activation, and hallucination scores; no effect was seen on the BPRS withdrawal, anxiety-depression, or hostility-suspicion scores (Deakin et al., 2008). Euphoria was not affected by lamotrigine, but dissociation scores were attenuated, with a significant reduction in CADSS total, derealization, and depersonalization scores. Most of ketamine’s neural effects were attenuated by lamotrigine.
Prefrontal theta cordance was reduced by ketamine (0.54 mg/kg IV, 30-min) at 10 and 30 min in a DBRCT crossover study of 20 healthy people (Horacek et al., 2010). The association of theta cordance with psychotomimetic (BPRS) effects was tested by comparing subjects who had a reduction in cordance (n=16 at 10 min, n=15 at 30 min) with those who did not (n=4 at 10 min, n=5 at 30 min). BPRS scores were higher in non-reducers, both for total score and for the withdrawal, thought disturbance-hallucinations, and activation subscales. Positive emotions were more common in reducers at 10 min and 30 min, whereas the groups did not differ on negative subjective effect scores. Because there were very few non-reducers, these results must be interpreted with caution.
Positive and negative emotion ratings increased, with 11 subjects reporting a change in positive emotions and five reporting a change in negative emotions (Horacek et al., 2010). At 10 min, higher ketamine and norketamine serum levels correlated with larger reductions in prefrontal theta cordance, but no correlation was observed at 30 min; also, there was no difference in ketamine and norketamine serum levels between theta cordance reducers and non-reducers. Because positive emotions were more common among reducers, a reduction in theta cordance could hypothetically be involved in the mood-elevating and antidepressant effects of ketamine.
In a resting state fMRI (rsfMRI) DBRCT crossover study of 19 healthy people, S-ketamine (0.25 mg/kg IV, 45-min) reduced functional connectivity of the default mode network (DMN) to the dorsal nexus (DN), pregenual ACC (pgACC), and the medial PFC (mPFC) one day after administration (Scheidegger et al., 2012). The posterior cingulate cortex (PCC) was used as the representative seed region for the DMN. Looking beyond the DMN, there was no change in functional connectivity between the PFC and amygdala nor a change in connectivity to the cognitive control network (CCN). Previous studies have shown the DN has elevated connectivity to the DMN in depression, suggesting ketamine-induced reduction of functional connectivity between the DMN and DN at 24 h could be relevant to its antidepressant effects. There was no correlation between acute psychotomimetic effects and changes in functional connectivity.
Global brain connectivity (GBC) increased in 22 healthy people tested after the start of a ketamine infusion (0.23 mg/kg IV bolus, then 0.58 mg/kg/h for 45-min) and in 12 people who were tested after the infusion (Driesen et al., 2013). Increased connectivity in some regions correlated with its positive and negative psychotomimetic effects, but there was no correlation between the number of voxels exhibiting increased GBC and psychotomimetic effects. Six clusters of increased connectivity were positively associated with positive psychotomimetic symptoms: the right insula, right planum temporale, bilateral pulvinar nuclei, left lingual gyrus, and the anterior cerebellar vermis.
Only one cluster correlated with negative symptoms (e.g. social withdrawal), specifically with increased GBC in the dorsal and medial anterior striatum and the thalamus correlating with reduced negative symptoms (Driesen et al., 2013). There were no regional relationships between GBC and cognitive symptoms (e.g. thought disorder and impaired concentration). Some of the regions with elevated connectivity contribute to interoceptive and exteroceptive processing, which could explain some of the psychotomimetic, dissociative, and perception altering effects.
Connectivity within the DMN is disrupted by ketamine at an anesthetic dose, particularly with corticocortical connections, not thalamocortical connections, as shown by Bonhomme et al. (2016) in a study of 14 healthy people who received ketamine at dosages producing light sedation and unresponsiveness. Ketamine caused propofol-like disruption of frontoparietal DMN connectivity and anticorrelated activity between the DMN and other regions. When the effect was increased from light sedation (plasma = 0.75 μg/mL) to deep sedation (plasma = 2 μg/mL), DMN connectivity was greatly impaired and there were no identifiable DMN clusters at the deepest level of sedation. Ketamine minimally affected executive control networks, the auditory network, the sensorimotor network, and the visual network.
Participants reported strange dreams during the infusion, including experiences of flying, seeing a bright light, a sense of body distortion, a sensation of dying, feeling imprisoned, and feeling wellbeing, joy, and peace (Bonhomme et al., 2016). When participants were moved at the end of the experiment, they often experienced nausea, which could be controlled using ondansetron and alizapride.
In an RCT of 53 healthy people, ketamine’s effects were assessed using fMRI and 1H-MRS (Javitt et al., 2018); 34 received ketamine and 19 received placebo. Ketamine was administered with an initial 0.23 mg/kg IV bolus followed by 0.58 mg/kg/h for 30-min and 0.29 mg/kg/h for 29 min. BOLD signal increased in the dorsal midcingulate cortex and in general, most of the cortex was activated by ketamine, except for the OFC. The largest increases were in the dorsal ACC and anterior insula. Analysis with 1H-MRS showed a moderate increase in Glx (glutamate + glutamine) in the mPFC exclusively during the first 15 min; that change was absent during the remaining 45 min of the infusion, resulting in a nonsignificant effect overall.
Between the placebo and ketamine groups, BOLD signal increases correlated with changes in BPRS, CADSS, POMS, and Psychotomimetic States Inventory (PSI) scores (Javitt et al., 2018). There was no correlation between BOLD response and subjective effect scores within the ketamine group, and there was no correlation between 1H-MRS Glx levels and subjective effects. Consistent with the large effect on fMRI results, BOLD response significantly differentiated the two groups, with a sensitivity of 85% and specificity of 90%, correctly identifying most of the ketamine participants.
3.5 History
General History of Ketamine
Phencyclidine (PCP; CI-395) was synthesized by the medicinal chemist Victor Harold Maddox in March 1956, who then provided it to researchers at the pharmaceutical company Parke Davis (Domino, 2010). Early studies found PCP possessed unique characteristics, such as causing a cataleptic-like state or drunkenness in animals. Dr. Edward Domino was encouraged to study PCP by Parke Davis pharmacology consultant Dr. Maurice Seevers. Dr. Domino demonstrated that it had anesthetic and delirium-like effects.
Its anesthetic efficacy in monkeys justified entering clinical development, with much of the work taking place in Michigan, including at Detroit Receiving Hospital. The initial human studies found it was distinct from classic sedative-type anesthetics it that it did not cause respiratory and cardiovascular depression (Maddox et al., 1965). Rather, it increased systolic blood pressure (SBP) by an average of 26 mmHg and diastolic blood pressure (DBP) by 19 mmHg at an ideal dose of 0.25 mg/kg (IV), and no arrhythmias occurred (Greifenstein et al., 1958). Those properties were accompanied by low toxicity overall, giving it an acceptable safety profile with the notable exception that it often caused an extended period of delirium/psychosis when its deep anesthetic effects wore off. At higher doses (0.5-1 mg/kg IV), PCP produced agitation and sometimes rigidity, catatonia, and seizures, but at appropriate doses it was largely benign aside from the psychotomimetic and hallucinatory emergence phenomena.
A study of PCP administered to 64 people demonstrated quick induction of anesthesia and analgesia without other anesthetics being required, depending on the operation (Greifenstein et al., 1958). Some patients responded to auditory stimuli, but they did not remember experiencing pain or anything else about the surgery.
Because of its good anesthetic effects and overall cardiovascular and respiratory stability, it entered clinical use under the name Sernyl and was adopted for anesthesia during surgeries primarily in the late 1950s (Hashimoto, 2019). Its postoperative deliriant and psychotomimetic effects, which could be coupled with agitation, proved to be a problem and it was determined not to be an acceptable anesthetic in humans, eventually resulting in the development of ketamine.
Dr. Carl Bratton, Parke Davis’ Head of Pharmaceutical Development, believed a short-acting derivative of PCP would be useful in anesthesia since PCP’s primary limitation was that its psychological effects lasted hours longer than an operation (Domino, 2010). After Dr. Bratton approved the synthesis of new PCP-like molecules, Calvin Lee Stevens, a professor of organic chemistry at Wayne State University in Detroit, made a series of PCP derivatives. One of those derivatives was ketamine (CI-581), which had positive effects and a good safety profile in animals, based on studies by Parke Davis (McCarthy et al., 1965). Parke Davis’s Head of Clinical Pharmacology, Dr. Alex Lane, moved the drug into clinical testing and was helped by Dr. Domino.
Dr. Domino, along with the anesthesiologist Dr. Guenter Corssen, administered the first intravenous dose of ketamine to a human on August 3, 1964. They studied subanesthetic and anesthetic doses to determine its dose-response profile; Dr. Domino characterized their findings with the substance as “remarkable” (Domino, 2010). Ketamine, like PCP, caused dissociation, hypertension, and tachycardia (Domino et al., 1965). Side effects occurred in one-third of volunteers and although most people reported unusual experiences like a sensation of floating in outer space or a loss of sensation in their arms and legs, ketamine mostly did not produce PCP-type emergence delirium. Its effects varied depending on the person, causing apprehension and aggression in some, while others became withdrawn (Domino et al., 1965).
Parke Davis proceeded carefully with ketamine due to concerns that it could end up being too similar to PCP, which would keep it from being approved for medical use. A psychiatrist associated with Parke Davis classified its post-anesthesia effects as similar to diethyl ether, not PCP (Domino, 2010). In reality, the qualitative effects of ketamine were similar to PCP, but the difference in its duration, intensity, and propensity to cause outright delirium did make ketamine a far superior anesthetic.
Because both drugs were unlike other medications, it was unclear how to describe ketamine’s subjective properties. Dr. Domino and his colleagues initially described the effects as “dreaming,” a term that had also been applied to PCP (Domino, 2010). Eventually, Dr. Domino mentioned how participants became “disconnected” from their environment while speaking with his wife Toni, who came up with the term “dissociative anesthetic.” To this day, that is how ketamine is classified and “dissociative” has become the standard classification for novel ketamine-like drugs.
Ketamine was approved by the FDA in the United States for use as a short-acting anesthetic in 1970 (Tyler et al., 2017). One its early use cases was as an anesthetic for American soldiers during the Vietnam War (Domino, 2010). Its use expanded into recreational settings shortly after its introduction, although it would not be scheduled as a controlled drug in the US until 1999, when it was placed in Schedule III.
Although its emergence phenomena were not as problematic as with PCP, they were still undesirable for clinicians and patients. Working with Dr. Elmer Zsigmond, Dr. Domino studied the interaction of ketamine with sedatives and found diazepam could be given alongside ketamine to reduce its emergence effects (Domino, 2010). Diazepam was replaced by midazolam and propofol over time because of their shorter durations.
Ketamine in Psychiatry
Once ketamine and PCP became recreational drugs, Dr. Domino observed multiple cases in the late 1970s and early 1980s in which users suffered from preexisting depression and experienced relief with ketamine (Domino, 2010). One woman claimed she received better antidepressant effects from ketamine and PCP than from her prescribed antidepressants, although the effects were transient and she had to repeatedly use them to maintain the benefits. Because there was no reason to suspect ketamine was suitable for clinical use as an antidepressant at the time, Dr. Domino recommended she stop that “bizarre practice.”
In addition to there being little reason to suspect either drug had useful antidepressant properties, PCP and ketamine were known to be psychotomimetic and PCP was under active investigation as a model for schizophrenia (Domino, 2010). As such, Dr. Domino believed it was wholly unwise to use them for psychiatric disorders. But as would become clear in the 1990s and 2000s, ketamine has genuine antidepressant effects and Dr. Domino may have been one of the first doctors to learn of them, even if he did not know it at the time.
Dr. Robert Berman and colleagues at Yale University conducted the first DBRCT of ketamine for depression in 2000. Ketamine was shown to produce rapid antidepressant effects that persisted for at least three days (Berman et al., 2000). In the 20 years since that trial, many studies have investigated the effects of ketamine in unipolar depression (particularly TRD) and bipolar depression, and a smaller number of promising studies have been published on its effects in obsessive-compulsive disorder (OCD), anxiety disorders, and posttraumatic stress disorder (PTSD).
Its efficacy in TRD has generated a lot of interest in the field of psychiatric medicine. Dr. Thomas Insel, former director of the National Institute of Mental Health (NIMH), said in 2014 that intravenous ketamine “might be the most important breakthrough in antidepressant treatment in decades.” Like many in the field, Insel cautioned that it was not yet ready for “broad use in the clinic” because too little is known about its safety and efficacy.
The view that ketamine is indeed one of the greatest breakthroughs in depression treatment in decades, but that people should proceed cautiously in using it is very common. Although it works for a large portion of people, the benefits of a single dose almost always fade or disappear within 3-7 days. Therefore, if ketamine is used by itself, repeated infusions are necessary. A series of 6-12 infusions may offer weeks or a few months of relief, but it is not known to be effective much longer than that. Needing to repeatedly use a medication is not inherently a problem—standard antidepressants are taken daily, of course—but ketamine is far from ideal if it is going to be given frequently.
While a single dose is nearly innocuous, the long-term safety of dozens of infusions is unknown, which is concerning since ketamine and related drugs are associated with neurotoxicity, cognitive impairment, and urological toxicity with heavy and/or chronic use. Furthermore, ketamine is distinct from standard antidepressants in that its acute effects are impairing and abusable, meaning it is difficult to imagine a scenario in which it could be widely prescribed for at-home use. If a patient must spend a few hours visiting a clinic every week to receive their treatment, that is more disruptive than taking a typical medication each day. For a severely treatment-resistant person it is clearly worthwhile to deal with that disruption, but it is not ideal.
To address these problems, pharmaceutical companies are actively developing other glutamatergic drugs to replicate ketamine’s antidepressant effects while reducing its abuse potential, impairment, and safety concerns.
Section 4: Racemic Ketamine For Mood Disorders
4.1 Introduction
Since the publication of the first DBRCT of ketamine for depression in 2000, in which it produced very positive results, there have been dozens of controlled trials and case reports documenting the effects of ketamine and its enantiomers in MDD and bipolar disorder (BD). As usual, there are outlier studies, but the research overall supports ketamine’s efficacy. Because the 3-7 days of relief typically provided by a single dose is not adequate for the average patient, repeated infusions or other methods of extending the effect have been a major area of interest, resulting in the development of a variety of drugs intended to replicate or extend ketamine’s benefits while improving tolerability.
A meta-analysis of nine studies evaluating single-dose ketamine in 234 patients, along with five studies on other glutamatergic investigatory drugs (CP-101,606; AZD6765; and GLYX-13), reported that ketamine was effective within 40-60 min, its effects peaked on Day 1, and the benefits persisted for 5-8 days (Kishimoto et al., 2016). The non-ketamine drugs had smaller and less consistent effects. When pooled together, they only had a significant effect on Days 5-8 and when the analysis was repeated just with the three AZD6765 studies, there was no significant effect at any time.
The response rate for ketamine was 43% at 40-60 min, 59% at 230-240 min, and 34% at Day 7; by comparison, placebo had a 0% response rate at 40-60 min, 2% at 230-240 min, and 8% at Day 7 (Kishimoto et al., 2016). Ketamine also had a larger remission rate beginning within 80 min: 18% at 80 min, 34% at Day 1, and 20% by Days 3-5; those remission rates were significantly superior to placebo, which only caused remission in 0-2% of patients. Although ketamine increased psychotic (BPRS) ratings at 40-60 min, those scores were lower compared to placebo on Day 3, and mania (YMRS) scores were lower with ketamine at all timepoints through Day 14 except for 40-60 min.
Similarly, a meta-analysis of seven DBRCTs with 149 MDD patients and 34 BD patients by McGirr et al. (2015) reported a significant effect of single-dose ketamine at 24 h, Day 3, and Day 7. MDD patients had a superior response compared to BD patients. There were transient dissociative (CADSS) and psychotomimetic (BPRS) effects, but there were no severe psychotic symptoms and the only instance of mania was in a BD patient who received placebo, not ketamine. The NNTs (number needed to treat) were as follows: At 24 h, 5 for remission and 3 for response; at Day 3, 6 for remission and 3 for response; and at Day 7, 6 for remission and 5 for response.
4.2 MDD and MDD+BD Research
Supporting Studies (Racemate)
Single Infusion
Berman et al. (2000) conducted the first DBRCT of ketamine for depression, treating nine unmedicated patients (MDD=8, BD=1), seven of whom completed both infusions. They received a 40-min infusion of 0.5 mg/kg ketamine compared with placebo. Over the following three-day period, ketamine-treated patients had a significant reduction in depression, with a mean HDRS score reduction of 14, compared to 0 with placebo. Based on HDRS ratings, 4/8 (50%) had a response during the 3-day follow-up, compared with 1/8 with placebo. Interestingly, the effects continued to improve during the follow-up period such that mean depression scores on Day 3 were lower than on Day 1 or at 240 min.
On specific symptoms. ketamine improved depressed mood, suicidality, hopelessness, and worthlessness; placebo did not improve ratings on any symptom scale (Berman et al., 2000). There was also a reduction in depression as assessed by the BDI, with a mean score change of 29.5 to 16.8 shortly after the infusion, compared to an increase from 23.0 to 25.2 with placebo. Nearly every patient lost the antidepressant effect within 1-2 weeks; one patient maintained a large benefit for at least two weeks, with the HDRS scores: 41 at baseline, 7 on Day 3, and 15 on Day 14.
During the acute infusion period, psychotic (BPRS) symptoms increased, particularly positive symptoms, and ketamine increased VAS ratings for ‘High,’ which returned to baseline by 110 min (Berman et al., 2000). BPRS, VAS-High, and HDRS scores did not correlate.
Weak evidence of a positive effect of ketamine exposure during surgery was reported in a study of 70 antidepressant-medicated MDD patients and 25 control patients undergoing orthopedic surgery (Kudoh et al., 2002). MDD patients were randomized into two groups: Group A (n=35) was anesthetized with propofol, fentanyl, and ketamine (1 mg/kg), while those in Group B (n=25) were only given propofol and fentanyl. Both groups were maintained using isoflurane plus nitrous oxide. Group C (control patients) received the same anesthesia as Group A. All patients received diclofenac every 6 h after the surgery for pain relief.
No patient in Group A had more than a 5 point increase on the HDRS, whereas 23% of Group B patients did (Kudoh et al., 2002). On Day 1, the mean HDRS increased from 12.3 to 14.4 in Group B, yet it was reduced from 12.7 to 9.9 in Group B, and depression ratings were essentially stable in control patients (4.2 to 4.8). Ketamine was significantly superior on Day 1, but not Day 3. On specific HDRS items, Group A had a reduction in depressed mood, suicidality, somatic anxiety, and hypochondriasis compared to Group B. Postoperative confusion was not elevated in ketamine-treated patients, occurring in 14% of Group A patients compared with 23% in Group B and 0% in Group C. One patient in Group A and two in Group B had an arrhythmia. Two patients each in Groups A and B developed hypotension (SBP < 70 mmHg). Postoperative pain was significantly lower in ketamine patients at 8 and 24 h, but not during the following four days.
Zarate et al. (2006) studied ketamine (0.5 mg/kg IV, 40-min) in 18 unmedicated TRD patients in a DBRCT crossover trial. It was superior to placebo at 110 min and that benefit remained significant in the following week. The effect size was large at 24 h (Cohen’s d = 1.46), decreasing to a moderate-to-large effect at Day 7 (d = 0.68). Because one of the patients initially treated with placebo withdrew due a medical illness, the analysis was conducted with the 17 people who received ketamine. Among those patients, the 24 h response rate was 71% with ketamine versus 0% with placebo, and the remission rate was 29% vs. 0%. Six (35%) maintained their response for at least one week and two still had a response two weeks later. Ketamine only increased BPRS positive scores at 40 min, at which time YMRS mania scores were also higher. Most adverse effects subsided within 80 min and no patient had derealization or depersonalization beyond 110 min.
In an open-label trial of 26 unmedicated TRD patients, ketamine (0.5 mg/kg IV, 40-min) was provided alongside placebo or lamotrigine (300 mg) (Mathew et al., 2010). At 24 h, the response rate was 65% and 54% had a response at Day 3. Day 3 responders entered a 32-day DBRCT of flexible-dose riluzole continuation therapy compared with placebo. The trial was stopped for futility because an interim analysis showed the relapse rate was higher among riluzole patients (80% vs. 50% with placebo).
Lamotrigine pretreatment did not block the transient side effects of ketamine or improve its antidepressant effect (Mathew et al., 2010). Response usually occurred within 240 min, with 46% exhibiting a response at 120 min and 62% at 240 min; response at 240 min was highly predictive of 24 h outcome. At 24 h, all 10 items on the MADRS had improved, with the greatest improvement in lassitude. There were no frank positive psychotic symptoms and 89% of patients had negligible BPRS positive scores.
Duncan et al. (2013) reported their findings from an open-label study of 30 unmedicated TRD patients treated with one infusion of ketamine (0.5 mg/kg IV, 40-min). Because this study was part of a larger trial evaluating riluzole as an add-on to ketamine, 19 patients received riluzole and 11 received placebo 4-6 hours after the infusion. There was a significant improvement with ketamine and no difference between the riluzole and non-riluzole groups, so they were pooled. Depressive symptoms (MADRS) were reduced at all timepoints. At 230 min, there were 13 responders and 17 nonresponders, giving a 43% response rate. Depressive symptoms were improved on Days 1 and 2 as well.
In one of the largest RCTs of ketamine for TRD, Murrough et al. (2013) compared ketamine (n=47) with midazolam (n=25) in unmedicated patients. Ketamine was administered as a single infusion (0.5 mg/kg IV, 40-min), as was midazolam (0.045 mg/kg IV, 40-min); midazolam was used instead of saline to improve blinding because it lacks antidepressant properties but is acutely psychoactive. Ketamine produced a superior response at 24 h, with a smaller mean MADRS score compared to midazolam (14.77 vs. 22.72). The response rate at 24 h favored ketamine (64% vs. 28%; NNT = 2.8).
Depression worsened in the following days, and by Day 7, ketamine had no significant effect on QIDS-SR ratings or response rate, but there were some signs of efficacy, e.g. a greater likelihood of scoring ≤2 on the CGI-Severity measure (Murrough et al., 2013). By Day 7, 21/30 ketamine responders and 4/7 midazolam responders still met response criteria.
Dissociative symptoms were reported by 17% of patients during the infusion, with effects like feeling outside of one’s body or altered time perception; those effects resolved by 2 h post-infusion (Murrough et al., 2013). No patient had severe psychotic symptoms (e.g. paranoia, hallucinations, delusions, thought disorder). Usually the blood pressure change was mild and transient, but the infusion had to be stopped in two patients because of hemodynamic effects: one person had hypertension (187/91 mmHg) that did not respond to β-blocker therapy, so the infusion was terminated after 30 min, producing BP normalization within 10 min; and in the other case, there was transient but pronounced hypotension and bradycardia that resolved without issue. Ketamine-induced adverse effects were not substantially different from midazolam, except for dissociative effects.
Hu et al. (2016) conducted a DBRCT with 30 depressed patients (56% had TRD) who were randomized to a combination of ketamine+escitalopram or escitalopram+placebo for four weeks. 15 patients received escitalopram (10 mg/d) and the other 15 patients received a single ketamine infusion of 0.5 mg/kg (IV, 40-min) plus daily escitalopram. Ketamine-treated patients had a larger response rate at Week 4 (92% vs. 57%). They also differed in their time to relapse, with an average of 27 days in the ketamine group and 6.4 days in the escitalopram-only group. In the subgroup of TRD patients, there was a 33% response rate without ketamine and an 89% response rate with it. The remission rate at Week 4 was 14% without ketamine and 77% with ketamine, and the average time to relapse was longer with ketamine (27 vs. 14 days).
Multiple Infusions
Fewer studies have looked at the effects of multiple infusions, but those that have indicate a series of infusions may increase the duration of efficacy.
Liebrenz et al. (2009) reported the successful treatment of TRD in a case report of a severely depressed male (HDRS = 36; BDI = 26). Along with TRD, he had alcohol and benzodiazepine dependence. He was tapered off his existing medication and received two open-label infusions (0.5 mg/kg IV, 50-min) over a 6-week period. There was a large reduction in depressive symptoms after the first infusion, with a peak effect on Day 2, at which time his HDRS had decreased by 57% and his BDI score was 65% smaller. Ketamine’s effect declined during the week and he was back to baseline when assessed on Day 35. The second infusion was effective, but the effects were smaller (-43% on HDRS; -35% on BDI); he returned to baseline within one week.
One of the first studies to use a series of infusions was an open-label trial of unmedicated TRD patients (n=10) who received six infusions (0.5 mg/kg IV, 40-min) over a 12-day period so long as they responded to the first dose (aan het Rot et al., 2010). These patients were part of an earlier single-dose open-label trial in which they had responded and then relapsed (Mathew et al., 2009); the mean time between trials was 311 days. With the first infusion, 9/10 had a response and continued with responders. The mean MADRS score for responders after the first infusion fell from 34 at baseline to 17 at 2 h and 7 at 24 h.
Four hours after the sixth infusion, 100% had a response and 89% experienced remission, with a mean MADRS of 5.0 (aan het Rot et al., 2010). After the last infusion, 8/9 relapsed in an average of 19 days; one person relapsed in the first week, whereas another was depression-free for >3 months. BPRS positive scores increased during the first infusion and returned to baseline within two hours; no patient reported distressing psychotic symptoms. Dissociation scores (CADSS) increased from 1.0 at baseline to a peak of 14.9 at 40 min, followed by a return to baseline within two hours. There was no evidence of persistent adverse cognitive effects, such as impaired memory or concentration. MADRS at 24 h did not correlate with peak BPRS positive scores or CADSS score.
Two patients had brief hypertensive episodes and transient tachycardia during the first infusion; both issues resolved within 5 minutes post-infusion (aan het Rot et al., 2010). One patient with tachycardia during the first infusion continued to experience the same with each subsequent infusion (max HR = 124 bpm) and despite having a normal baseline ECG and a negative cardiac history, they had asymptomatic premature ventricular contractions during two infusions, resolving within two hours. One patient had transient bradycardia (HR = 50-55 bpm) during the first infusion and the same occurred during most of the other infusions, but the effect never lasted more than two hours. A patient who presented with low DBP at baseline (107/48 mmHg) had a reduction in BP (80/55 mmHg) ~13 h after the first infusion, which remained low for at least 24 h; they also had mild transient hypotension during two other infusions. All of the hemodynamic effects were manageable. Two patients who entered the study with medication-controlled hypertension did not have hypertension during the infusions.
Blier et al. (2012) published a case report of a female with a long history of treatment-resistance who had previously responded to one ECT course but not a second. While she continued with her existing medications, ketamine (0.5 mg/kg IV, 40-min) significantly reduced her depressive symptoms. Her dysphoria and anxiety were reduced by 50% within an hour, but the benefits only lasted 36 hours. The duration was extended to 3-4 days when she received two infusions a few days apart. Her treatment course ultimately involved 41 infusions; she responded better to a 0.5 mg/kg infusion than a 0.2 mg/kg bolus. Despite the large number of injections over a period of months, there was no evidence of cognitive impairment (assessed with MoCA). Ketamine did not fully alleviate her depression, but her symptoms were reduced to the point that she could be more active instead of spending much of her time crying in bed.
Murrough et al. (2012) performed an open-label study of up to six ketamine infusions (0.5 mg/kg IV, 40-min) given to 24 unmedicated TRD patients over a 12-day period. 22 received at least two injections and 21 received all six doses. Data from the first 10 patients in this study was previously reported (aan het Rot, 2010). Within two hours of the first infusion, the mean MADRS declined from 32 at baseline to 13. There was a clear separation between responders and nonresponders by 4 h (MADRS: 10.4 vs. 19.0) and the difference between responders and nonresponders was large at 24 h (MADRS: 8.4 vs. 18.8). Ketamine was not entirely ineffective in the nonresponders, as they still had a reduction in MADRS scores for sadness, inner tension, pessimistic thoughts, and suicidal thoughts at 2 h. Responders tended to improve after their initial 2 h depression reduction, whereas the nonresponders worsened over time. 94% of people who responded at 24 h had a response by 4 h, while 29% of nonresponders had a response at 4 h.
The response rate was 71% one day after the sixth infusion (Murrough et al., 2012). Responders and nonresponders did not differ on psychotomimetic, dissociative, or ‘high’ ratings. After the last dose, responders were followed for 83 days or until relapse; 14 patients did not receive psychotropic medications during the follow-up period, while three enrolled in an RCT of venlafaxine XR for relapse prevention (two randomized to venlafaxine, one to placebo). Responders had a median time to relapse of 18 days, with a large range of 4 to >83 days; the follow-up results were similar between medicated and unmedicated patients. Four patients did not relapse during the 83-day follow-up period.
Ketamine produced mild but significant increases in psychotomimetic (BPRS) and dissociative (CADSS) symptoms, which resolved by 240 min (Murrough et al., 2012). Ketamine also caused transient mood elevation, as assessed by YMRS and VAS-High scores. No patient had clinically significant psychotomimetic effects (e.g. paranoia, delusions, hallucinations). The most common acute side effects were feeling strange or unreal (58%), abnormal sensations (54%), blurred vision (50%), and drowsiness (46%). There were no serious adverse events and 67% had no clinically significant vital sign changes. 33% had elevated BP and/or HR based on the study criteria of SBP/DBP of >180/100 mmHg, or HR of >110 bpm. One patient had hypertension (max: 180/115 mmHg) that did not respond to antihypertensive medication, so the infusion was discontinued, which quickly resolved the increase.
In a small open-label study of 10 TRD patients treated with twice weekly ketamine (0.5 mg/kg IV, 100-min) for two weeks, there was a highly significant reduction in depression, with an 80% response rate and a 50% remission rate (Rasmussen et al., 2013). Most patients were maintained on existing pharmacotherapies and some had dose increases during the study. Remission occurred with one dose (n=1), two doses (n=3), or four doses (n=1). Remission was sustained in 2/5 patients during a 4-week follow-up period; those two patients were on antidepressants. Suicidality was reduced and the improvement correlated with general depression reduction.
No patient experienced mania, with only occasional reports of racing thoughts, circumstantial speech, or sleeplessness (Rasmussen et al., 2013). There were no instances of arrhythmia or significant BP elevation nor did any patient require respiratory support. At 2 h and 24 h, psychotomimetic (BPRS) scores were slightly lower than baseline, which at least indicates there were no persistent psychotomimetic effects.
An open-label trial of 14 TRD patients treated with six infusions (0.5 mg/kg IV, 40-min) over a 12-day period reported a large 92% response rate after the final infusion and a 67% remission rate, whereas the first infusion produced a response in 25% and remission in 8%, demonstrating the utility of multiple infusions (Shiroma et al., 2014). Because there were two dropouts, the analysis only used the 12 completers. Patients could continue a stable dose of their existing medication and those who had a response by the final infusion were followed for four weeks or until relapse; four patients had a history of suicide attempts. Among the 11 responders, five maintained their response during the 4-week follow-up and the mean time to relapse was 16 days (range: 7-28 days).
There was a small increase in psychotomimetic (BPRS) and dissociative (CADSS) effects, returning to baseline by 120 min (Shiroma et al., 2014). Those side effects did not correlate with the antidepressant effect of ketamine. No patient had an arrhythmia or needed respiratory support, but one normotensive patient experienced hypertension (180/92 mmHg) that was rapidly responsive to labetalol treatment. One patient with GERD had nausea/vomiting during the second infusion; ondansetron was successfully given prophylactically for the rest of the infusions. One patient dropped out after the first infusion because of reduced energy and increased irritability, and another dropped out after the second infusion due to dissatisfaction with the therapeutic effect of ketamine.
An RCT of 18 MDD patients who were randomized to repeated ketamine (n=9) or ECT (n=9) found ketamine was superior at multiple timepoints (Ghasemi et al., 2014). The ECT group received three sessions separated by 48 h, while the ketamine group received three infusions (0.5 mg/kg IV, 45-min) separated by 48 h; patients could continue with their existing medications. Ketamine-treated patients had fewer symptoms after the first and second treatments, and at 72 h post-treatment. After the first treatment, 44% responded with ketamine, but only 11% responded with ECT, and ketamine reduced symptoms by 42% on average, compared with a 12% reduction from ECT.
Most of ketamine’s symptom reduction occurred with the first infusion and that effect was maintained and slightly improved upon through the subsequent two infusions, persisting for at least one week after the last treatment (Ghasemi et al., 2014). In contrast, ECT was relatively ineffective in the first session but continued to improve with each treatment. In those who responded to the treatments, both produced effects that persisted for one week. There were no significant HR or BP changes with either treatment, with only a transient, non-clinically significant increase in HR and SBP in three patients during the second and third doses of ketamine.
Comparatively small efficacy ratings were reported in an open-label study of 28 treatment-resistant MDD or BD patients who were assigned either to Group A that received ketamine (0.5 mg/kg IV, 40-min) once weekly for three weeks (n=15) or to Group B that received two infusions each week for three weeks (n=13) (Diamond et al., 2014). Patients could continue with their existing medications during the study. In Group A, 80% completed all infusions, compared with 62% in Group B; two discontinued because of acute adverse effects and five stopped due to a lack of efficacy and increased anxiety. At 6 h, 11% of patients had a response, while on Day 21 the response rate was 29% (33% of completers), specifically with 33% of patients in Group A and 23% in Group B.
Suicidal ideation was reduced by 61% within six hours of a single infusion and the reduction persisted to Day 21 in responders, whereas it was absent in nonresponders (Diamond et al., 2014). In Group A, one person withdrew because of a panic attack during the first infusion that included tachycardia and tachypnea, and another withdrew after the second infusion because of elevated anxiety and a perceived lack of benefit. In Group B, two withdrew after the first infusion: one had a brief vasovagal episode (BP 77/47; HR 45) with reduced consciousness that resolved within an hour, and the other patient had increased anxiety, worsening of mood, and increased suicidality, which was attributed to a loss of hope because of the inefficacy of the treatment. Two others withdrew after four infusions and one after the fifth infusion because of increased anxiety and a lack of benefit.
The side effects that did not result in treatment discontinuation included include one patient with rapid mood cycling after three infusions, resulting in a mild hypomanic episode with racing thoughts and reduced sleep, which was alleviated by an increase in their antipsychotic medication (Diamond et al., 2014). Three other patients, all of whom were prone to fluctuating levels of suicidality at baseline, had mood instability during the trial that included worsening of mood and increased suicidality. Most patients experienced fatigue during the afternoon after the infusions and some patients had mild headaches.
Zheng et al. (2018) conducted a large open-label study of unipolar (n=77) and bipolar depression (n=20) patients treated with six ketamine infusions (0.5 mg/kg IV, 40-min) over 12 days while they remained on a stable existing medication. 24 h after the first infusion, the response rate was only 14% and the remission rate was 9%, but by 24 h after the final infusion, response was present in 68% and the remission rate was 51%. The average time to response was 11 days. There were improvements in depressive symptoms, anxiety, and suicidality within four hours; those benefits were maintained through the trial. Response 24 h after the first infusion positively correlated with response after the sixth. Nonresponders also had a rapid reduction in suicidality.
Dose-Response
Most studies have used a 40-min IV infusion of 0.5 mg/kg, but few studies have examined its dose-response profile. Those that have suggest other doses may be effective, but if the dose is too low, the effects become smaller and shorter-lasting.
This topic was explored by Xu et al. (2015) in a meta-analysis of nine trials (201 subjects). Six trials used low-dose ketamine (0.5 mg/kg IV) and three tested very low-dose ketamine (50 mg intranasal; 0.1-0.4 mg/kg IV; and 0.1-0.5 mg/kg IV, IM, and SC). Two studies exclusively used treatment-resistant BD patients, while the others studied treatment-resistant unipolar depression. The reduction in depression on Day 3 was smaller in the very low-dose trials and among BD patients versus MDD patients.
Pooled results of the very low-dose trials found no significant response or remission effect on Days 1, 3, or 7, and the effect that existed was smaller than with 0.5 mg/kg IV (Xu et al., 2015). Very low-dose intranasal ketamine produced the least acute side effects, where very low-dose ketamine given as an IV push dose caused more substantial side effects. Seven study authors provided the meta-analysis authors with unpublished data on suicidality; although baseline scores were low, suicidality was reduced post-ketamine on Days 1 and 3, but not by Day 7.
Lai and colleagues (2014) tested the dose-response profile of ketamine in four TRD patients. They could continue a stable pharmacotherapy during the study if it had failed to provide adequate relief. The effects of 0.1-0.4 mg/kg were tested using a rapid (5-min) IV infusion, with a randomly inserted placebo dose for comparison. Within the first 72 h, 3/4 had a response with at least one treatment: two responded at 0.1 mg/kg and one had an apparent dose-dependent effect, showing a response at 0.4 mg/kg. Response lasted at least 24-48 h, but it was gone by Day 7.
The dissociative and psychotomimetic side effects seemed to be more dose-dependent, and the side effects varied by patient (Lai et al., 2014). Two people had transient tachycardia (up to 150 bpm) and one had hypertension (140/80 mmHg increasing to 195/105 mmHg) during the 0.4 mg/kg infusion; those problems resolved within 10 min. The side effects were generally mild and resolved within four hours, including headaches, dizziness, lightheadedness, perioral numbness, and nausea. By 4 h, orientation and reaction times were not significantly affected.
Su et al. (2017) reported a small, nonsignificantly superior response with 0.5 mg/kg (IV, 40-min) compared with 0.2 mg/kg in a DBRCT of 71 Taiwanese MDD patients. Response, which was defined as a ≥50% reduction in HDRS on at least two days between Days 2 and 5, occurred in 12% of placebo patients, 39% of 0.2 mg/kg patients, and 46% of 0.5 mg/kg patients. The two doses were not significantly different from each other, but only 0.5 mg/kg was significantly superior to placebo.
Animal Research
Supporting Studies
Repeated ketamine (10 mg/kg IP) administration in CUMS-susceptible male rats was effective and it reduced the time lag for citalopram-induced antidepressant effects (Zhang et al., 2015). CUMS reduced bodyweight and sucrose preference. Over a three-week period of ketamine exposure, bodyweight increased, sucrose preference increased, and FST immobility was reduced when animals received ketamine weekly, every three days (E3D), daily for one week, or daily for three days. The greatest effects occurred with weekly and E3D dosing, suggesting weekly administration may be ideal. When ketamine was administered daily for three or seven days, its effects typically only lasted until Day 14, whereas weekly and E3D dosing produced benefits for the full three weeks.
Citalopram alone did not increase sucrose preference and bodyweight until Day 14, and FST immobility was not reduced until Day 21 (Zhang et al., 2015). In contrast, the combination of weekly ketamine with daily citalopram led to improvements from Day 7 onward, although efficacy was also observed by Day 7 with ketamine-only, impeding the detection of a unique effect with the combination.
4.3 Bipolar Disorder (BD)
Supporting Research
DiazGranados and colleagues at the US NIMH studied the effects of ketamine (0.5 mg/kg IV, 40-min) as an adjunctive therapy for BD patients (n=18) maintained on lithium or valproate in a DBRCT crossover trial (DiazGranados et al., 2010). Ketamine reduced depression by 40 min and its effect lasted until at least Day 3, with the largest effect (Cohen’s d = 0.80) on Day 2. 71% of patients responded at some point during the trial with ketamine compared with only 6% of patients after placebo. No benefit was seen on Days 7, 10, or 14.
Consistent with those results, Zarate et al. (2012) reported antidepressant effects with ketamine (0.5 mg/kg IV, 40-min) in a DBRCT crossover trial of 15 BD patients maintained on lithium or valproate. Patients had a mean lifetime history of 10 antidepressant trials, 40% had attempted suicide, 73% had a lifetime history of anxiety, 60% had a lifetime history of alcohol abuse or dependence, 40% had a history of substance abuse or dependence, and 33% had a history of psychosis.
The improvements appeared by 40 min and lasted to Day 3, but it did not have a significant effect on Day 7 (Zarate et al., 2012). 73% (11/15) completed both infusions, with three dropping out after a ketamine infusion and one dropped out after placebo; two of the post-ketamine dropouts had a brief response that was lost by Days 3 or 7. Ketamine produced its greatest effect at 40 min (Cohen’s d = 0.85), which reduced by Day 1 (d=0.70) and Day 2 (d=0.65). There was no difference in baseline MADRS before the first and second infusions, which were separated by two weeks, indicating a lack of carryover effect.
Ketamine improved 8/10 MADRS symptoms, only failing to improve reduced appetite and decreased sleep (Zarate et al., 2012). The response rate was 64% at 40 min, 50% at 230 min, and 43% on Day 1. In total, 79% of ketamine-treated patients responded at some point, but 0% of placebo-treated patients had a response. The median time to relapse was two days, although two patients maintained their response for at least one week, a third responded for at least 10 days, and one person had a response until at least Day 14. Ketamine only increased dissociation (CADSS) at 40 min and it did not significantly affect mania (YMRS) or psychotic symptom (BPRS) ratings.
Cusin et al. (2012) described two cases in which long-term ketamine was effective in treatment-resistant BD. The first patient was a female with bipolar II disorder and ADHD who had not responded to 12 antidepressant trials or ECT during her last depressive episode; at present she was taking venlafaxine, lamotrigine, and methylphenidate. Ketamine was initiated at 0.5 mg/kg (IV, 40-min) twice weekly. Her symptoms were reduced after the third infusion, including passive suicidal ideation, anhedonia, and fatigue; her symptoms fully returned within ten days of the fifth infusion.
Ketamine was not effective orally (210 mg three times per week) or intranasally (three times per week) for three weeks (Cusin et al., 2012). Subsequently the patient received 32 mg IM and 50 mg IM, producing complete depression remission. She was administered 50 mg IM every four days for five months, at which time she partially relapsed. The dose was increased to 70 mg, which maintained complete remission for four months; the main side effects were irritability, nightmares, and dissociation.
The second patient was a female with bipolar II disorder, ADHD, chronic depression, and suicidal ideation that did not respond to mood stabilizers, SNRIs, TCAs, or MAOIs (Cusin et al., 2012). She was prescribed lamotrigine, levothyroxine, pregabalin, armodafinil, and oxcarbazepine. Oral ketamine (150 mg) three times per week was ineffective and 100 mg IM was intolerable because of dissociation. Her symptoms improved within one week of switching to 50 mg IM every three days. She partially relapsed after six months and bupropion was added; although she was not in remission, her suicidality was gone and she had been able to work since receiving ketamine therapy. The side effects were headache and irritability.
Depression was reduced for at least one week after a single infusion (0.5 mg/kg IV, 40-min) in an uncontrolled study of 18 BD patients concurrently receiving mood stabilizers (Permoda-Osip et al., 2015). The mean HDRS fell from 24 at baseline to 13 on Day 3 and 12 on Day 7; 44% (8/18) were responders on Day 7.
An analysis of data from two RCTs (36 patients) in which ketamine (0.5 mg/kg IV, 40-min) was administered for BD found anxious and non-anxious patients had the same antidepressant response even though the presence of anxiety correlated with higher baseline MADRS and HDRS depression scores (Ionescu et al., 2015). Patients could continue a stable dose of lithium or valproate during the study; 21 had anxious depression and 15 did not.
Specific Symptoms
Ketamine reduced fatigue from 40 min until Day 14, except for Day 7, in two DBRCT crossover trials (36 patients) of ketamine (0.5 mg/kg IV, 40-min) for treatment-resistant bipolar I or II (Saligan et al., 2016). The largest effect was on Day 2 (d=0.58) and the benefit remained significant after controlling for general depression reduction. Ketamine had a 65% response rate for fatigue symptoms compared with 10% among placebo-treated patients.
D-Cycloserine Add-On
Treatment-resistant BD patients (n=8) who received open-label ketamine (0.5 mg/kg IV, 60-min) followed by eight weeks of D-cycloserine plus pyridoxine while on existing mood stabilizers and/or benzodiazepines had a large reduction in depressive symptoms (Kantrowitz et al., 2015). D-cycloserine was titrated from 250 mg to 1000 mg/d over a 3-week period. 7/8 patients completed the study, with four meeting remission criteria at Week 8; there was a significant improvement at all timepoints other than Week 2. Ketamine had a large effect on Day 1 (d=2.0) and at Week 8 (d=1.1); the acute response to ketamine was predictive of response at the end of the study.
4.4 Alternative Routes of Administration (Non-IV)
Aside from IV administration, ketamine may be effective when given via the intranasal (IN), IM, sublingual, subcutaneous (SC), and oral routes. There is not enough research to definitively say which is best, but IV and oral likely sit on opposite sides of the spectrum for reliable, acute efficacy. IV administration maximizes ketamine exposure and consistently shows fast-acting effects, whereas oral administration produces much more norketamine due to greater first-pass metabolism, it may require multiple days of use before the benefits begin, and total efficacy could be reduced. That said, daily oral ketamine seems to be tolerable and it could therefore be used to provide longer-lasting benefits than IV dosing so long as it is safe.
Loo et al. (2016) studied the effects of ketamine (0.1-0.5 mg/kg) in TRD when administered via IV (n=4; 5-min infusion), IM (n=5), and SC (n=6) compared with midazolam, which was randomly inserted as a control. Patients could remain on an inadequate antidepressant medication during the study. 12/15 patients had a response at least once during the trial, with response/remission rates of 75% with IV, 60% with IM, and 100% with SC; only two people responded to midazolam. Response rate increased from placebo up to 0.4 mg/kg, suggestive of dose-dependency. Of the 12 responders, 10 were followed until relapse and the mean time to relapse was 23 days overall: 9 days with IV, 12 days with IM, and 35 days with SC (only 16 days after excluding an outlier who responded for >150 days).
The strongest dissociative effects were with IV, but there was no significant effect of ROA on dissociation at 40 min or 240 min (Loo et al., 2016). HR and BP transiently increased, with a peak incidence at 5-10 min with IV versus 10-15 min with SC and IM; the smallest cardiovascular effects were with SC. There was a linear correlation between plasma concentration and dosage, and ketamine concentration correlated with CADSS at 40 min; plasma levels were similar between IM and IV at 0.4-0.5 mg/kg, and SC produced similar concentrations to IM. Because it was easier to administer via the SC route, which also provided a good response rate and reduced side effect burden, that could be a preferable route to use.
Oral
Two hospice patients with depression and anxiety experienced relief with a single oral dose of 0.5 mg/kg; because ketamine also alleviated pain, that could have mediated some of the benefits (Irwin and Iglewicz, 2010). In the first patient, pain was reduced through Day 4 and their HDRS-17 fell from 32 at baseline to 16 one-hour post-ketamine, remaining greatly reduced on Day 15 (HDRS = 10). It produced either no change or improvements in BPRS (psychotic), YMRS (mania), and MMSE (cognitive dysfunction) ratings. Her suicidal thoughts subsided within two hours and she experienced other benefits over time, including: more engaged, resumption of prior interests, more alert during the day, appetite improvement, feeling relaxed, and reduced anxiety (HADS-A: 16 at baseline —> 6 on Day 15). Her depressive symptoms returned after a month and she was given a second dose, which did not work; however, she was also experiencing delirium at the time. Her symptoms later subsided while on bupropion, gabapentin, and analgesics.
The second patient had been psychiatrically healthy, but severe depression developed while he was undergoing treatment for terminal cancer; he experienced suicidal thinking, anhedonia, social withdrawal, anxiety, and panic (Irwin and Iglewicz, 2010). His anxiety and pain improved within an hour of ketamine administration and he felt calmer. Depressive symptoms mostly improved by Days 3-4 and they continued to improve through the following week. Among the benefits were increased appetite and increased socialization; it did not negatively affect cognition. He was only assessed until Day 13 because of a decline in health, but his wife reported that his thoughts about death shifted from suicidal (i.e. wanting to be dead) to acceptance of death—he died within the next two weeks. His HDRS-17 had decreased from 28 at baseline to 13 on Day 8 and his HADS-A reduced from 13 to 2.
Irwin et al. (2013) proceeded to study the effects of nightly ketamine (0.5 mg/kg/d) in 14 hospice patients during a 28-day open-label study. It produced significant benefits after Day 14, with a mean time to response of 8.6 days for anxiety and 14.4 days for depression. Ketamine did not affect pain, functional status, quality of life ratings, suicide risk, or cognition. Four patients withdrew after Day 14 because of inefficacy and two others dropped out for unrelated reasons, leaving eight patients who could be analyzed. 8/8 patients had a ≥30% reduction in both anxiety and depression scores, and clinical exams confirmed the improvements on Days 14 and 28. There were mild increases in diarrhea (n=1), insomnia (n=1), and difficulty sitting still (n=1). The neurological and psychiatric side effects started to decline by Day 7 and the GI symptoms were reduced by Day 21.
In a 6-week DBRCT of oral ketamine (150 mg/d; n=20) versus diclofenac (n=20) in chronic pain patients with mild to moderate depression, ketamine was superior at both HDRS measurement points (Weeks 3 and 6) (Jafarinia et al., 2016). The response rates were 40% vs. 15% at Week 3 and 60% vs. 15% at Week 6.
Oral ketamine was compared with placebo in a 6-week DBRCT of 81 patients during the introduction of daily sertraline, with ketamine being given at 50 mg/d; patients were evaluated at 2, 4, and 6 weeks (Arabzadeh et al., 2018). It produced significant benefits at all timepoints. Early improvement (≥20% HDRS change within two weeks) was more common in the ketamine group (85% vs. 43%) and the Week 6 response rate favored ketamine (85% vs. 58%), but there were no significant differences on remission. Ketamine did not cause dissociation.
Intranasal
The 24 h response rate was 44% (8/18) among 20 TRD patients treated in a DBRCT crossover trial of single-dose ketamine (50 mg IN) (Lapidus et al., 2014). Patients could continue a stable existing pharmacotherapy. Ketamine was more effective than placebo at 24 h (44% vs. 6% response; NNT=2.6), based on MADRS ratings. Ketamine was at least numerically superior for seven days, although the difference was only significant at 40 min, 240 min, 24 h, and 48 h, not on Days 3 or 7.
Dissociative and psychotomimetic effects occurred shortly after administration, but they did not correlate with the antidepressant effects (Lapidus et al., 2014). The most common adverse effects during the four hours after infusion were feeling strange or unreal, poor memory, and weakness or fatigue. No clinically significant cardiovascular effects occurred. SBP increased by 7.6 mmHg at 40 min and only four people had an SBP >130 mmHg, compared to three people with placebo. The mean plasma concentration of ketamine was 72 ng/mL at 20 min and 84 ng/mL at 40 min; those levels were smaller than a typical 0.5 mg/kg IV infusion.
Intramuscular
If it is similarly effective to IV dosing, IM could be more widely implemented at a lower cost because it does not require an IV infusion pump, is easier to administer, and is an option for people with poor venous access.
An open-label dose-response study of two female TRD patients reported that 0.5 mg/kg (IM) only reduced depression by 10-20%, but larger effects occurred with 0.7 and 1.0 mg/kg, with one patient reaching remission (Glue et al., 2011). The adverse effects were similar to those of IV use, such as lightheadedness, sedation, and dissociation.
Chilukuri et al. (2014) conducted a randomized open-label trial with 27 MDD patients comparing IV ketamine (0.5 mg/kg, 40-min) with IM ketamine (0.25 and 0.5 mg/kg); there were nine subjects in each group and they were studied for three days. HDRS scores were reduced 59% at 2 h in the IV group, compared with 60% and 57% with 0.5 and 0.25 mg/kg (IM), respectively—relief was sustained for at least three days. This suggests ketamine is similarly effective with both routes and 0.25 mg/kg may be adequate for nearly maximizing the benefits.
Sublingual/Transmucosal
In a case series of 26 treatment-resistant MDD or BD patients treated with low-dose sublingual ketamine who were allowed to continue an existing pharmacotherapy, 77% (20/26) experienced response or remission for depression and improvements in mood instability, cognitive impairment, and poor sleep; three patients had a partial response and three did not respond (Lara et al., 2013). Treatment consisted of a liquid containing 10 mg, which was kept in the mouth for 5 min before being swallowed; the dose could be changed as needed. Patients received ketamine at variable frequencies from weekly to 2-3x per week.
Of the 11 who were studied acutely after the first dose, 8/11 had a clear response after the first dose and some described the effects as “sensational” or “incredible” and used phrases like “I am back to life again” (Lara et al., 2013). Ketamine increased confidence without causing an altered sensation like that of stimulants and patients frequently reported sleep improvement. It did not cause manic, psychotic, or dissociative symptoms, although two BD patients experienced transient agitation. It often produced mild lightheadedness that resolved within 30 min and became less prominent with subsequent doses.
A similar response rate was reported in a retrospective chart review of 17 TRD patients treated with 0.5 or 1 mg/kg, which was placed on the tongue and held in the mouth as long as possible (Nguyen et al., 2015). These patients were on other medications (SNRIs, stimulants, benzodiazepines, SSRIs, etc) and they were in a treatment program that included group therapy sessions, including within one hour of ketamine administration. 76% (13/17) responded, usually in less than 24 h; those who failed to respond within the first day typically did not go on to benefit.
Ketamine was initially given every other week, but the frequency increased to every 7-10 days for some patients due to a loss of efficacy within two weeks (Nguyen et al., 2015). Seven responders dosed every two weeks, three dosed every 10 days, and one dosed weekly; frequency information was not available for the other responders. No serious adverse effects occurred and no patient discontinued because of adverse effects. There were only mild side effects, namely light headache (n=1) and slight dizziness (n=1).
4.5 Use in Psychotic Patients
Because of ketamine’s acute psychotomimetic and dissociative effects, and therefore its hypothetical ability to worsen preexisting psychosis, most studies have excluded patients who have psychotic symptoms or disorders. While it is reasonable to think ketamine is riskier in this population, the risk may be overstated. A number of studies have found its effects in depressed patients with psychosis are similar to its effects in non-psychotic populations, with only transient psychotomimetic side effects.
Supporting Studies
Ribeiro et al. (2016) reported the successful treatment of depression in two females with comorbid psychotic symptoms. The first patient presented with a long history of depression and psychotic symptoms. She experienced auditory hallucinations, paranoid delusions, and suicidality; her medications included venlafaxine, ziprasidone, clonazepam, and zolpidem. Because she had not adequately responded to her medications, ketamine was tried. It caused mild dissociative symptoms, fatigue, and headache during the infusion. Afterwards, she did not have depression, and her auditory hallucinations and paranoia subsided within hours of the infusion. She was ultimately treated with three infusions and transitioned from venlafaxine to an MAOI to prevent relapse; her HDRS scores declined from 19 to 9 during treatment.
The second patient had a history of severe schizoaffective disorder, depressive symptoms, and suicidality; she was withdrawn, whispering, and catatonic at presentation (Ribeiro et al., 2016). Her medications included clozapine, lamotrigine, and lithium. In the past, she had engaged in self-mutilation, overdosed on medications, and set her house on fire during a psychotic episode involving hallucinatory commands. At one point she was given ketamine for anesthesia during ECT, which produced a large antidepressant effect despite no seizure being induced. Because of her past response to ketamine and the severity of her persistent symptoms, ketamine was administered for depression. Her mood improved post-infusion and her psychotic symptoms disappeared. After five infusions and medication changes, she was discharged without suicidal ideation and with improved mood; her HDRS declined from 29 to 8 during treatment.
An analysis of three RCTs of ketamine (0.5 mg/kg IV, 40-min) involving 69 patients, 12 of whom had a history of psychosis, found ketamine was safe and efficacious in the subgroup with a history of psychosis (Pennybaker et al., 2017). Two studies were on BD patients concurrently treated with lithium or valproate and the third mostly consisted of unmedicated MDD patients; most patients with a history of psychosis had BD with psychotic features (n=10). The psychosis subgroup had greater dissociation (CADSS) at 40 min, but not higher psychotomimetic (BPRS) scores, and the difference was not observed at other timepoints. Although ketamine was effective in both groups, with depression relief in the psychosis subgroup for up to three days, the effect size was smaller in the psychosis subgroup (Cohen’s d = 0.35 vs. 1.17). The groups did not differ on other relevant factors like baseline MADRS, baseline BPRS-positive, BMI, family history of alcohol use disorder, or prior suicide attempts.
A case series of S-ketamine infusion (0.5 mg/kg IV, 40-min) for depression found it was effective in 3/4 people with concurrent psychotic symptoms (Ajub and Lacerda, 2018). The first patient—who also had alcohol dependence—experienced mild dissociative symptoms, but within two hours the side effects had resolved and she reported substantial improvement in her depressive and psychotic symptoms; her suicidality was absent at 24 h. Two weeks later, the patient reported mild depressive symptoms but not psychotic symptoms. The second patient (MDD; SAD) presented with psychotic symptoms, insomnia, depressed mood, intense anxiety, auditory hallucinations, ideas of reference, and blunted affect. S-ketamine caused intense dissociative symptoms that resolved by 2 h; her psychotic and depressive symptoms were remitted at 24 h and she remained free of both for four weeks.
The third patient (BD; alcohol abuse) presented with suicidality, severe depression, auditory hallucinations, ideas of reference, cognitive deficits, and dysphoria; she was hospitalized because of her high imminent suicide risk. One day after infusion, her suicidality, depression, and psychotic symptoms were completely remitted; she continued with her existing medications and remained free of psychiatric problems for at least two weeks. In the fourth patient (schizoaffective disorder), S-ketamine caused moderate dissociation but no intensification of psychotic symptoms, although it also did not alleviate depression at 24 h. Three additional once-weekly infusions also failed to have an effect.
4.6 Effects on Specific Symptoms
Psychiatric medicine is shifting towards a focus on symptom clusters and their neurobiological causes instead of broad diagnostic categories. A disorder like depression comes in multiple forms that may call for different kinds of treatment. For a long time, physicians have treated symptom clusters and identified subtypes of depression without much evidence to guide their choices, but there is now an effort to carefully identify symptoms (e.g. cognitive deficits, anhedonia, social aversion) that involve unique neurocircuitry dysregulation. This effort is exemplified by the Research Domain Criteria (RDoC) project from the US NIMH (Insel et al., 2010).
While current diagnostic tools like the American Psychiatric Association’s Diagnostics and Statistical Manual of Mental Disorders (DSM) and the World Health Organization’s International Classification of Diseases (ICD) help with reliably diagnosing patients with a broadly applicable disorder, those diagnoses can overlook the nuances of an individual patient’s condition, yet treatment based on those nuances could improve outcomes (Insel et al., 2010). Widely accessible tools (e.g. fMRI, electrophysiology) can be used to study the underlying physiology of disorders and their symptoms. The RDoC project is centered around using those tools to identify biomarkers that will assist with detecting and treating subtypes of common psychiatric conditions.
As with most psychiatric research, the ketamine literature predominantly focuses on categories like MDD and bipolar disorder, but some researchers have looked at its effects on specific problems like anhedonia and suicidality, aiming to determine which patients will benefit the most from ketamine.
Anhedonia
Anhedonia is a common symptom of depression and other psychiatric disorders that involves a deficit in the ability to experience pleasure from rewarding stimuli, impaired motivational activity (resulting in less effort to seek out rewarding stimuli), and/or deficits in reward learning and reactivity to positive stimuli; as a result, positive reinforcement is impaired (Pizzagalli, 2014). It may involve reduced dopaminergic transmission in reward and motivational circuits caused by a detrimental effect of chronic stress on mesocorticolimbic pathways, i.e. dopaminergic pathways projecting from the ventral tegmental area (VTA) to the NAc/striatum and PFC.
Supporting Research
A DBRCT crossover trial of treatment-resistant BD patients (n=36) comparing ketamine (0.5 mg/kg IV, 40-min) with placebo reported an anti-anhedonic effect of ketamine during the two-week period after administration (Lally et al., 2014). They had failed to respond to open-label lithium or valproate, but they were maintained on those during the trial. The benefits first appeared around 40 min and persisted for up to two weeks in some patients. Although anhedonia positively correlated with overall depression (MADRS total score), an anti-anhedonic effect of ketamine remained after controlling for other symptoms.
The anti-anhedonic effect was larger in patients with a family history of alcohol use disorder, but there was no correlation with a patient’s personal history of alcohol abuse, dependence, or illicit substance abuse (Lally et al., 2014). A subgroup (n=21) was studied using PET imaging >2 h after infusion. Patients with the greatest increase in rCMRGlu (regional glucose metabolism, i.e. indicative of activity) in the ventral striatum tended to have the greatest reduction in anhedonia at 230 min, but change in overall depression predicted the effect, so there was no specific connection to anhedonia. Reduced anhedonia at 230 min correlated with increased glucose metabolism in the dorsal ACC (dACC), extending into the pregenual cingulate/callosal region and right dlPFC; however, the correlation with dACC metabolism was not significant when assessing anhedonia on Day 14. There was a trend-level correlation between anhedonia reduction and increased metabolism in the fusiform gyrus, extending into the claustrum and putamen. Glucose metabolism in the OFC was not correlated with anhedonia reduction.
In an open-label study of 52 unmedicated TRD patients receiving ketamine (0.5 mg/kg IV, 40-min) followed by randomization to riluzole (n=26) or placebo (n=26), anhedonia was significantly or trend-level reduced at 40, 80, 120, and 230 min, as well as Days 1-3 (Lally et al., 2015). Some patients (n=20) underwent FDG-PET analysis at baseline and >2 h post-ketamine. Anhedonia, which was significant at baseline in 87% of patients, did not correlate with baseline cerebral metabolic rate of glucose, but there was a correlation between reduced anhedonia and increased glucose metabolism in the dACC, which remained significant after controlling for overall depression reduction.
Reduced anhedonia correlated with increased glucose metabolism in the right hippocampus and with reduced metabolism in the right OFC and left inferior frontal gyrus (IFG); after controlling for total depression score, the hippocampal and OFC correlations remained significant or were at a trend-level, but the IFG correlation was lost (Lally et al., 2015). Subjects with a family history of alcohol use disorder had a superior anti-anhedonic response, whereas no correlation was observed for comorbid anxiety or for depression subtype (melancholic vs. others). Although riluzole was initiated 4-6 h after the infusion, it did not affect ketamine’s anti-anhedonic property or depression in general.
Relevant Research (Non-Ketamine)
Anhedonia may be a predictor of more persistent depression, as shown in a prospective study of a general population sample in the Netherlands (n=7076) that was assessed at baseline and 12 months later (Spijker et al., 2001). 305 (4.3%) met the criteria for major depression in the preceding six months at baseline. 24% were lost to attrition during the follow-up period. For the 223 who were available a year later, 28% again met the criteria for major depression in the past six months. The likelihood of persistent depression correlated with anhedonia (n=146; OR=3.32), psychomotor agitation or retardation (n=102; OR=2.08), and early morning awakening (n=84; OR=2.35).
In another analysis of adults in the Netherlands (n=7076), 586 (8.2%) had a depressive disorder; 97/586 (17%) reported suicidal ideation and 19 (3.2%) had attempted suicide in the past two years (Spijker et al., 2010). Depression was defined as simultaneously having at least two depressive symptoms on the CIDI, with at least one key symptom. Anhedonia (OR=2.00) correlated with the risk of suicidality, as did comorbid anxiety (OR=2.46), feelings of worthlessness (OR=1.99), and longer duration of depression (OR=2.86).
Out of five symptom dimensions (reported depressed mood, anhedonia, somatic symptoms, morbid thoughts, and observed depression), anhedonia was the strongest predictor of nonresponse in patients (n=334) who did not respond to SSRI treatment and were subsequently switched to another medication or medication + CBT during a 24-week treatment period (McMakin et al., 2012). Anhedonia was also associated with a longer time to remission and more days with depression, unlike the other symptom dimensions.
Vrieze et al. (2013) used a computerized probabilistic reward task in which reward learning performance negatively correlates with self-reported anhedonia to evaluate 79 MDD inpatients and 63 healthy controls. 56% (44/79) of patients had high anhedonic symptoms (SHAPS >7) at baseline. After eight weeks of treatment, reward learning was reduced among patients compared to control subjects and high-anhedonia patients had reduced reward learning compared to low-anhedonia patients. Deficits in reward learning at baseline greatly increased the likelihood of persistent MDD eight weeks later (OR=7.84).
Suicidality
Suicidal ideation is a major symptom of depressive disorders and other psychiatric conditions. It is often reduced by standard therapies, but it can take weeks for those medications to work and in the meantime, some patients may be at a high risk of self-harm. Few medications are associated with a specific anti-suicidal effect, among which are lithium, clozapine, and ECT.
Ketamine seems to have an anti-suicidal effect that is clinically meaningful, rapid, and long-lasting (often 3-7 days). It is a very promising therapy for this application given the current lack of treatment options.
An analysis of patient data (n=167) from 10 studies comparing ketamine with saline or midazolam in people with suicidal ideation found a rapid decrease in suicidality within one day (Wilkinson et al., 2018). The mean baseline MADRS was 33 and 48% of patients were receiving pharmacotherapy. Patients were studied for one week and experienced a moderate-to-large effect (Cohen’s d = 0.48-0.85) at all timepoints; an anti-suicidal effect was present after controlling for general depression reduction, although suicidality and depression scores were highly correlated. The mean MADRS score for ketamine patients fell from 34 at baseline to 20 on Day 1 and 22 on Day 7, whereas control patients minimally changed: 33 at baseline, 29 on Day 1, and 28 on Day 7.
Current Treatment Options
With daily administration for months or years, lithium appears to have an anti-suicidal effect in bipolar and unipolar depression patients that may be independent of general mood improvement (Baldessarini et al., 2006; Kovacsics et al., 2009).
Supporting Research
Suicidality was reduced after a single infusion of ketamine (0.5 mg/kg IV, 40-min) in 26 unmedicated TRD patients, nine of whom then enrolled in a trial of six infusions over a 12-day period (Price et al., 2009). The suicidality item of MADRS (MADRS-SI; 0 to 6 scale) was reduced by 2.1 points on average and eight (62%) had a rating of 0 or 1 one day after the infusion; however, the anti-suicidal effect was nonsignificant after controlling for general depression reduction. At 24 h, three patients had fleeting suicidal thoughts (MADRS-SI = 2 or 3) and two remained at ≥4 on the MADRS-SI.
A subsample of 12 patients also completed the Implicit Association Test (IAT), which measures implicit suicidality (Price et al., 2009). Scores on the Escape=Me component (i.e. wanting an escape) correlated with MADRS-SI, but not with general depression severity, and baseline Death=Me associations (i.e. wanting death) did not correlate with other measures. At 24 h, there was only a reduction in Escape=Me associations, which had a trend-level correlation with MADRS-SI, but not with general depression ratings. The repeated-dose regimen caused a similar reduction in suicidality (MADRS-SI). Ketamine maintained its efficacy through the 12-day treatment period, with a mean reduction of 2.9 points on the MADRS-SI from baseline to Day 12.
DiazGranados et al. (2010) reported a rapid reduction in suicidality among MDD patients (n=33) treated with open-label ketamine (0.5 mg/kg IV, 40-min) and the reduction was greatest in those with higher baseline suicidality. Patients with higher baseline suicidality on the SSI also had higher MADRS and HDRS total depression scores, higher MADRS and HDRS suicidality scores, higher HDRS anxiety, and a larger history of prior suicidal ideation and suicide attempts. Ketamine had a large effect at 40 min (Cohen’s d = 1.05) and a moderate effect at 230 min (d = 0.45). Patients with higher baseline suicidality (SSI >3) experienced larger effects at 40 min (d = 2.36) and 230 min (d = 1.27); the difference was even larger at 40 min (d = 3.13) and 230 min (d = 1.84) in those with baseline SSI >4. Similar patterns in ketamine’s effect were reported for total depression and anxiety scores, and for the BDI hopelessness score.
A significant anti-suicidal effect was reported in a DBRCT crossover trial of ketamine (0.5 mg/kg IV, 40-min) in 15 BD patients (Zarate et al., 2012). Suicidality was reduced from 40 min to Day 3.
An analysis of four studies involving treatment-resistant MDD (n=98) and BD (n=35) found an anti-suicidal effect from ketamine (0.5 mg/kg IV, 40-min) that was partially independent of changes in overall depression and anxiety (Ballard et al., 2014). 81 patients (61%) had suicidal ideation at baseline (i.e. HDRS-Suicide >0), 40% of whom had previously attempted suicide. 51 patients participated in a controlled crossover study and had a significant improvement post-ketamine. The reduction in suicidality correlated with depression reduction, but change in depression only accounted for a small portion (19%) of the effect. Ketamine increased ratings on the ‘wish to live’ factor of the SSI and reduced ‘wish to die.’ The greatest effect occurred at 40 min.
In a study of 57 unmedicated TRD patients who were treated in a DBRCT manner with ketamine (n=36; 0.5 mg/kg IV, 40-min) or midazolam (n=21), suicidal ideation at 24 h was reduced despite patients only having low-to-moderate suicidality at baseline (Price et al., 2014). 53% of ketamine patients scored 0 on three measures of explicit suicidal ideation compared with only 23% of midazolam patients and 7% of all patients at baseline. The reduction in explicit suicidality was mediated by a general reduction in depression (MADRS total) at 4 and 24 h. Unlike midazolam, ketamine also reduced implicit suicidal ideation (‘Escape=Me’ associations) at 24 h. Ketamine was most effective in patients with higher baseline suicidality, greater implicit suicidal ideation, and a history of suicide attempts. This study was limited by its exclusion of patients who were at imminent risk of suicide or needed hospitalization.
Ketamine reduced suicidal ideation more than midazolam in an RCT of 24 people with mood and anxiety disorders who had clinically significant suicidality at baseline, defined as MADRS-SI ≥4 (Murrough et al., 2015). 54% of patients had MDD, 29% had BD, and 12.5% had PTSD; baseline depression severity was moderate-to-severe and 63% had a history of suicide attempt. Most patients were on existing medications during the study. Ketamine did not show efficacy on the BSI at 24 h (the primary outcome measure), but it significantly reduced BSI scores at 48 h; the difference between groups was lost by Days 3 and 7. Ketamine was superior for reducing irritability and panic at 24 h; it did not have a larger effect than midazolam on mania, anxiety, or insomnia. During a 5-week follow-up, four patients were hospitalized for worsening depression or suicidality, which was not considered to be related to ketamine.
Vande Voort et al. (2016) studied the effects of six infusions (0.5 mg/kg IV, 100-min) over a two-week period in 12 treatment-resistant unipolar or BD patients who were hospitalized with suicidal ideation; those who remitted during the first phase of treatment entered a continuation phase with four weekly infusions followed by four weeks without treatment. 42% (5/12) remitted and 58% (7/12) responded during the first phase, though only seven patients completed that phase. Two stopped because of adverse effects, one stopped because of efficacy, and two stopped due to clinical worsening: specifically, one patient had treatment-emergent behavioral outburst with suicidal threat and one attempted suicide.
Four of the first-phase remitters reached remission with one infusion, while the other patient required three infusions. Suicidality was also significantly reduced during the first phase (Vande Voort et al., 2016). Further depression reduction occurred during the continuation phase in the five patients who continued therapy. 4/5 were no longer remitted during the post-ketamine follow-up, but they still met response criteria. The adverse effects included dissociation (n=9), dizziness (n=7), numbness or tingling in extremities (n=7), drowsiness (n=6), tearfulness/emotionality (n=4), and facial numbness (n=3). Almost every adverse effect subsided within two hours.
Bartoli et al. (2017) reviewed the results of five trials with 99 patients who had suicidality at baseline: 63 received an IV bolus (0.2 mg/kg) and 36 were given an infusion (0.5 mg/kg, 40-min). Ketamine reduced suicidality within 40 min and continued to have an effect at 4 h.
Suicidal ideation was reduced one day after ketamine (0.5 mg/kg IV, 40-min) compared to midazolam in an RCT of 80 MDD patients who had current suicidal ideation (SSI ≥4); 54% were on an existing antidepressant and the average baseline SSI was ~15 (Grunebaum et al., 2018). Ketamine reduced SSI scores 5 points more than midazolam at 24 h (Cohen’s d = 0.75), producing a response rate of 55% with ketamine and 30% with midazolam. Overall mood (POMS) improvement was greater on Day 1 with ketamine versus midazolam and change on that metric accounted for 34% of the SSI reduction. During an uncontrolled follow-up period consisting of additional pharmacotherapy, clinical improvement persisted for up to six weeks.
Ketamine was also associated with reduced suicidal desire and ideation scores on the SSI in patients who continued to have suicidal ideation, but it did not have a superior effect on the SSI planning subscale (Grunebaum et al., 2018). Worsening of suicidal ideation occurred in nine midazolam patients and two ketamine patients on Day 1. Ketamine nonsignificantly improved general depression ratings, with the following response rates (ketamine vs. midazolam): HDRS-17 (30% vs. 15%), HDRS-24 (25% vs. 15%), and BDI (36% vs. 17%).
Open-label ketamine was provided to 35 midazolam patients who did not remit, producing an 8-point reduction on the SSI on Day 1, a 7-point reduction on the HDRS-17, and a 10-point reduction on the HDRS-24 (Grunebaum et al., 2018). Ketamine transiently increased SBP by 15 mmHg vs. 4 mmHg with midazolam and DBP by 13 mmHg vs. 4 mmHg with midazolam. No suicides occurred during a six-week follow-up, but there were two suicides afterwards, one at six months and one at 26 months; both had received ketamine, which caused remission in one and no effect in the other.
Cognitive Functioning
Many patients with depressive disorders have impaired cognitive functioning and those impairments can persist after otherwise successful treatment. While excessive ketamine use can semi-persistently impair cognition and antidepressant doses acutely impair cognitive performance, ketamine appears to have either no effect or a positive effect on cognition in depressed patients after the acute effects subside.
In some of the studies on ketamine or other antidepressants, no placebo control was used, which means the retest effect could be responsible for apparent drug-mediated improvements.
Background
A review of 11 studies assessing cognitive performance in 500 remitted unipolar depression patients compared with 471 control subjects reported that performance was impaired in at least one test in most studies (Hasselbalch et al., 2011). In 2/3 studies that tested a hypothetical connection between persistent impairment and persistence of subclinical depressive symptoms, MADRS score correlated with performance in some tests and the cognitive impairment became nonsignificant after controlling for subclinical symptoms. Among the five studies that assessed the impact of the number of depressive episodes, three found no connection, while two reported worse performance in patients with more depressive episodes.
Likewise, Bora et al. (2012) performed a meta-analysis of 27 studies comparing non-depressed (euthymic) adult MDD patients (n=898) with healthy controls (n=997) and found that euthymic patients performed significantly worse on cognitive tests, usually with modest impairments. Euthymia was defined as remission for at least two months with a current HDRS <7 or MADRS<10 and ‘late-onset depression’ was defined as depression onset after 50-65 years old. Late-onset patients had greater deficits in verbal memory, information processing speed, and some executive functions. Differences between remitted patients and healthy controls were seen in the Stroop interference, Trail-Making Test parts A and B, digit span backwards, list learning, list recall, and animal naming tests, but not in the phonetic fluency, WCST perseveration, digit span forwards, and list recognition tests.
Cognitive function was still impaired during remission and after antidepressant discontinuation in MDD patients treated with escitalopram (n=36) or duloxetine (n=37) compared with control subjects (n=37) (Herrera-Guzman et al., 2010). Some functions improved over time in MDD patients, but overall, they had inferior performance during remission (while on antidepressants) in verbal and visual episodic memory, sustained attention, mnemonic and strategic aspects of working memory, and planning. The same pattern of impairment was present during a 24-week recovery phase after medication discontinuation.
Baune et al. (2010) found greater impairments in current medicated MDD patients (n=26) compared with those who had a past history of MDD (n=44) or healthy controls (n=206), although some deficits in immediate memory and attention persisted in people with a history of MDD. HDRS-17 scores were higher among current patients than former patients (18.0 vs. 6.8); most subjects in both groups were taking SSRIs or SNRIs. Compared with previously depressed patients, subjects with current depression had worse visuospatial/constructional and attention scores. There was no association between cognitive function scores and either quality of life (physical or mental) or activities of daily living.
Remitted MDD patients (n=88) scored worse than control subjects (n=50) on measures of attention and visuomotor speed, and on a test of executive function (Hasselbalch et al., 2012). Remission was defined as HDRS <8 and no depressive episode in the past two months. The mean HDRS score was only 2.8 and patients had been remitted for an average of 6.4 years; 63% were still prophylactically taking psychiatric medication. In contrast with some of the existing literature, there was no correlation between task performance and subclinical depressive symptoms.
Supporting Research
Diamond et al. (2014) reported generally positive cognitive effects in 28 treatment-resistant MDD or BD patients who received ketamine 1-2x per week for three weeks. Improvements in autobiographical memory and episodic memory were found, and subjective memory performance was neutral or improved. Two patients could only recall <78% of baseline items on autobiographical memory tests, which is comparable in magnitude to the deficit observed after ECT, but with a far smaller incidence. Furthermore, the single patient who had a decline in all measures of autobiographical memory had been withdrawn from the ketamine study and received ECT on the morning of their Day 21 assessment; the deficit cannot be clearly associated with ketamine.
As a result of depression reduction rather than a specific pro-cognitive effect of ketamine, performance in visual memory and working memory tests (simple and complex) improved in 15 TRD patients treated with six infusions (0.5 mg/kg IV, 40-min) over a 12-day period (Shiroma et al., 2014). Lower attention scores at baseline correlated with superior response.
Ketamine improved cognitive performance for three days in 18 BD patients who were concurrently taking mood stabilizers (Permoda-Osip et al., 2015). A single infusion (0.5 mg/kg IV, 40-min) was associated with improved performance on tasks pertaining to psychomotor speed, visual attention, task switching, and executive functions. Patients with worse baseline functioning had a greater effect. Change in depression on Days 3 or 7 did not correlate with the effects on cognition.
Neutral Research
There was no cognitive impairment in the days following ketamine treatment (0.5 mg/kg IV, 40-min) compared with midazolam in a DBRCT of 63 unmedicated TRD patients, 43 of whom received ketamine (Murrough et al., 2015). Cognitive performance improved on Day 7 in both groups, with no superior effect from ketamine. Improved performance was reported in tasks related to processing speed, verbal learning, and visual learning, whereas there was no improvement in working memory or reasoning. Slower baseline processing speed predicted depression reduction at 24 h in the ketamine group and ketamine responders had slower baseline processing speed versus nonresponders; baseline processing speed did not correlate with baseline depression severity.
4.7 Add-On Therapies
Riluzole
Ketamine’s antidepressant effect in unmedicated TRD patients was only nonsignificantly enhanced in those who received riluzole + ketamine (n=21) compared with ketamine-only (n=21); this DBRCT initially provided patients with open-label ketamine, which was then followed by randomization to riluzole or placebo 4-6 h after ketamine (Ibrahim et al., 2012). Riluzole began at 100 mg/d and could be adjusted to a max of 200 mg/d; the RCT portion of the study went for 28 days. Before randomization, 62% responded to ketamine. 58% of responders did not relapse in the first week, 38% had not relapsed by the end of the second week, and 27% (7/26) did not relapse for at least four weeks:
Depression was reduced from Day 1 through Day 28 with no difference between the placebo and riluzole groups (Ibrahim et al., 2012). The study completion rate was 64%. At the end of four weeks, 21% (3/14) of placebo patients had not relapsed compared with 33% (21%) of riluzole patients. Pooled together, patients scored an average of 6.0 points lower on MADRS at the end of the study versus baseline, with a large reduction in effect size over time, falling from a large effect (d=1.02) on Day 2 to a moderate effect (d=0.46) on Day 28. The mean time to relapse was 13 days (17 days with riluzole vs. 10 days with placebo). If the difference in relapse time was replicated in a larger sample, it could be clinically meaningful.
Cognitive Therapy
Cognitive therapy (e.g. CBT) after antidepressant treatment may reduce the risk of relapse and it also seems to have a smaller risk of relapse compared to pharmacotherapy in general. Therefore, it may be preferable to use cognitive therapy in place of continued pharmacotherapy, particularly considering MDD patients seem to prefer psychotherapy over medication and treatment adherence is improved when people receive their preferred treatment (Raue et al., 2009).
In a review of 13 studies with patients (n=728) who were successfully treated with pharmacotherapy and then given CBT versus control patients (n=682), CBT was associated with a reduced risk of relapse (Guidi et al., 2016). Patients assigned to CBT after medication discontinuation were less likely to relapse than patients who received clinical management or continued with their medication (RR=0.674), and CBT reduced the risk of relapse when added to ongoing antidepressant therapy compared with antidepressant-only treatment.
Upon cessation of therapy, depression treatment with cognitive therapy had a smaller risk of relapse than antidepressant treatment (Hollon et al., 2005). In a trial of 180 patients with moderate-to-severe depression who were treated with cognitive therapy or antidepressants, the response rate was 58% (n=104). Those who responded to cognitive therapy were less likely to relapse during the 12 months following the end of treatment compared to people withdrawn from antidepressants (31% vs. 76%) and they were no more likely to relapse than patients maintained on medication (31% vs. 47%).
Supporting Research
Wilkinson et al. (2017) reported a small 8-week relapse rate of 25% in an open-label, uncontrolled study of 16 TRD patients treated with four infusions (0.5 mg/kg IV, 40-min) over a two-week period followed by CBT for eight weeks (12 sessions total). 69% of patients had a history of hospitalization, 69% had melancholic depression, and 38% had received ECT; they continued with their existing pharmacotherapy during the trial. Ketamine produced a 50% (8/16) response rate and 44% (7/16) remitted during the first two weeks.
At the end of the CBT course, only 25% (2/8) of responders had relapsed, although longer-term follow-up showed that 5/8 responders relapsed in a median of 12 weeks post-ketamine (Wilkinson et al., 2017). Ketamine nonresponders did not benefit from CBT. Most ketamine responders showed a response after the first infusion (6/8), while the others responded after the fourth infusion. During the ketamine phase of the trial, working memory and visual memory improved after controlling for change in depression severity, although there was a negative effect on processing speed.
4.8 Ketamine With ECT
Some studies have reported a positive effect of ketamine anesthesia on the antidepressant effect of ECT, but the interaction is not consistent. A meta-analysis of five RCTs with 182 patients (MDD=65; BD=17) found no difference in remission (24% vs. 23%) or response (40% vs. 45%) with ketamine + ECT compared with ECT alone (McGirr et al., 2015). After removing one study in which no patient responded, the overall response and remission rates increased, but ketamine still did not have an effect: response in 53% vs. 58% and remission in 31% vs. 29% in ketamine-treated compared with non-ketamine patients. It was compared with anesthesia using propofol or thiopental.
Ketamine increased seizure duration and was associated with a greater risk of confusion/disorientation/delirium (OR=6.59), but most adverse effects did not differ between the groups, including hypertension and agitation. Two lithium-treated BD patients experienced an affective switch after ECT + ketamine compared with zero patients in the other group; one entered hypomania and one entered a mixed state (McGirr et al., 2015). In the two studies that assessed cognitive functioning, ketamine was associated with a beneficial effect on cognitive performance (MMSE) in one and had no effect in the other.
Supporting Research
The antidepressant effect of eight ECT sessions over a 4-week period in TRD inpatients was greater in patients who received ketamine (n=11) than in those who were given propofol (n=20), though depression was reduced in both groups and ketamine was only significantly superior to propofol after the second and fourth sessions, not the sixth and eighth (Okamoto et al., 2010). The mean anesthetic dose of ketamine was 0.86 mg/kg (IV) and patients could with existing pharmacotherapy during the study. Seizure duration tended to be longer with ketamine, but the difference was only significant in the first and sixth sessions.
Ketamine-treated patients had a higher rate of hypertension (55% vs. 20%) and hallucinations with a sense of fear post-awakening (27% vs. 0%), whereas propofol-treated patients had more angialgia, i.e. vascular-related pain (45% vs. 0%) (Okamoto et al., 2010). One person dropped out because of fear and hallucinations after an ECT session with ketamine. Because ketamine’s apparent effect was observed during the early ECT sessions, it may primarily accelerate depression relief in the beginning of treatment.
Kranaster et al. (2011) reported a beneficial effect of S-ketamine in a retrospective chart review of 42 treatment-resistant depression patients (primarily unipolar) treated with ECT; anesthesia was induced with S-ketamine (n=16) or thiopental (n=26). Patients who received ketamine needed fewer ECT sessions to complete treatment and they had lower HDRS scores. There was a slight improvement in cognition (assessed with MMSE) in ketamine patients and a slight decline in the thiopental group. There was no difference in motor response time or duration of EEG activity, but seizure concordance (a seizure quality parameter) was greater with ketamine.
Cardiovascular side effects were more common when ketamine was used, necessitating more BP control with urapidil (Kranaster et al., 2011). Two ketamine patients experienced cardiac side effects: one had intermittent atrial fibrillation and the other had ventricular extrasystoles in the context of preexisting intermittent ventricular extrasystoles. Those patients fully recovered. Postictal agitation occurred in four thiopental patients and no ketamine patients. One patient with a history of derealization experienced derealization and nightmares after ECT with ketamine anesthesia; those problems did not occur when thiopental was used.
Opposing Research
Ketamine did not enhance the effect of six ECT sessions delivered over a 2-week period in 18 depressed patients who were either given thiopental alone (n=9) or thiopental with ketamine (0.5 mg/kg IV) (Abdallah et al., 2012). Patients could continue their existing pharmacotherapy during the trial. Both groups had a reduction in depression and neither treatment was superior. The first session failed to improve HDRS-25 scores, but there was a significant effect 24-72 h after the sixth session, with a 13% response rate in the ketamine group versus a 38% response rate in the control group.
HDRS scores were reduced from 35 to 23 (baseline vs. post-sixth session) with ketamine, while the control group declined from 38 to 20 (Abdallah et al., 2012). There was a trend towards longer seizure duration in the ketamine group along with an increase in motor seizure duration. Interpretation of this study is complicated by the small sample size and differences between the groups: there were more MDD patients in the ketamine group (87% vs. 37%) and mood stabilizer use was higher in the control group (25% vs. 75%).
Neutral Research
ECT with ketamine anesthesia greatly reduced depression in a female patient with severe TRD; she was withdrawn from valproate and lamotrigine before the first session (Ostroff et al., 2005). Anesthesia was induced with ketamine (0.5 mg/kg IV) because of its putative protective effect against cognitive impairment post-ECT, which had been a problem for the patient after ECT in the past. Although no seizure occurred in the first session, she reported immediate mood improvement upon regaining consciousness. Her improved mood persisted into the next day, at which time she received a second ECT session. Again, ECT did not induce a seizure, but the antidepressant was even greater; she reported improvements in every symptom the following day, including interest, energy, motivation, mood, and appetite (Ostroff et al., 2005). The response persisted during a 5-day period after the second infusion, though with waning efficacy. She had a full tonic-clonic seizure during the third session and required three additional sessions to attain remission. Because ketamine was not compared with another treatment, it is impossible to assess ketamine’s contribution to the antidepressant effect.
Section 5: Ketamine For Other Psychiatric Disorders
5.1 Posttraumatic Stress Disorder (PTSD)
Supporting Research
A review of medical charts for 147 American soldiers who were treated for burns, had completed the PTSD Checklist-Military (PCL-M), and had at least one operation found a lower prevalence of PTSD in those who received ketamine during their surgery (n=119) compared with those who did not (n=28) (McGhee et al., 2008). The prevalence was 27% with ketamine and 46% without it despite ketamine-treated patients having, on average, more severe injuries, larger burns, longer ICU stays, and a greater number of surgical procedures. Unlike the weak correlation between PTSD and ketamine exposure, there was no correlation with morphine equivalent units administered during surgery, burn size, injury severity, ICU duration, or number of operations.
D’Andea and Sewell (2013) described the case of a 23-year-old veteran suffering from severe chronic PTSD and depression; he had a childhood history of trauma and abuse. During a 3-year period in which he was repeatedly hospitalized, he was treated at different times with fluoxetine, citalopram, escitalopram, bupropion, olanzapine, amitriptyline, valproate, venlafaxine, and risperidone; those drugs were either inadequate or intolerable. He also had insomnia that was resistant to zolpidem, quetiapine, trazodone, diphenhydramine, prazosin, and eszopiclone. Ketamine was initiated because he had become increasingly despondent, reported suicidal ideation, and his child was due to be born in the near future.
His existing medications were discontinued 24 h before ketamine (35 mg IV, 20-min) combined with propofol (D’Andrea and Sewell, 2013). After awakening, the only side effects were transient nystagmus and visual distortions. The antidepressant effect was large and fast-acting. His mental state drastically improved on the same day as the infusion and one week later his personality was improved, leading to improved functioning and interaction with others (e.g. he voluntarily enrolled in a birthing class). Ketamine fully resolved his anxiety, hyperarousal, and insomnia. The effect of ketamine wore off by Day 15 and he returned to his prior state within one day; at first, he experienced irritability, negative thinking, and anhedonia, which was followed by insomnia and violent dreams (D’Andrea and Sewell, 2013).
Ketamine was transiently effective in a DBRCT crossover trial of 41 unmedicated PTSD patients treated with one infusion (0.5 mg/kg IV, 40-min) compared with midazolam (Feder et al., 2014). The ketamine-first and midazolam-first patients had a mean past-month CAPS of 82.5 and 77.1, respectively. 29/41 patients completed both infusions; six noncompleters did not receive the second infusion due to a persistent CAPS <50 post-ketamine. PTSD symptoms (IES-R) were lower with ketamine at 24 h, but although some difference (8.7 points) persisted until Day 7, there was no longer a substantial difference in CAPS score between groups. Baseline MADRS and the presence of MDD did not affect IES-R score at 24 h. Ketamine had similar effects on the IES-R items for intrusion, avoidance, and hyperarousal symptoms.
By Week 2, seven ketamine responders still had a significant reduction in symptoms compared with only one midazolam responder (Feder et al., 2014). Ketamine did not cause significant psychotic or manic symptoms and its acute dissociative effect resolved by 120 min. Three patients needed β-blocker treatment during ketamine infusion because of an increase in SBP >180 or DBP >100. Patient-rated side effects during the first 24 h were similar between treatments, except for seemingly more nausea/vomiting, blurred vision, and restlessness post-ketamine: blurred vision (36% ketamine vs. 19% midazolam); dry mouth (21% vs. 16%); restlessness (23% vs. 10%); fatigue (21% vs. 23%); nausea/vomiting (21% vs. 3%); poor concentration (15% vs. 3%); and headache (13% vs. 13%).
Depression and PTSD symptoms were reduced in a study of 15 PTSD patients with comorbid TRD who received six open-label infusions (0.5 mg/kg IV, 40-min) over a 12-day period (Albott et al., 2018). Patients continued their existing medications during the trial. The mean baseline MADRS was 36 and the mean CAPS-5 was 40; most had PTSD from combat exposure or sexual assault. Ketamine produced PTSD remission in 80% and depression response in 93%, as assessed 24 h after the final infusion. Those with PTSD remission at the end of the infusion series (n=12) had a median time to relapse of 41 days and patients with depression response (n=14) had a median time to relapse of 20 days for depressive symptoms. Ketamine did not worsen PTSD in any patient.
PTSD remission was present in nine patients after the first infusion and in 12 after the second, whereas depression response was much more likely after the sixth infusion compared to the first (93% vs. 20%) (Albott et al., 2018). All 12 patients with PTSD remission had a reduction in depression, but there were three depression responders who did not experience PTSD remission. Six people remained in PTSD remission during 56 days of follow-up. Dissociative effects occurred after the start of each infusion, but there was no significant mood elevation or problematic psychotomimetic effects; the dissociative and psychotomimetic side effects did not increase in magnitude during the 6-infusion course. Three (20%) patients needed β-blocker treatment because of SBP >180 and/or DBP >100.
Opposing Research
Ketamine exposure during treatment for severe burns did not correlate with a smaller risk of PTSD 16-19 months later in a group of 30 male patients who were compared with 15 healthy controls (Winter and Irle, 2004). In every patient, the onset of PTSD was soon after the traumatic incident; no patient was on psychotropic medication. Patients with PTSD (n=15) and patients without PTSD (n=15) had smaller right hippocampal volume relative to controls and larger burn surface area correlated with smaller left hippocampal volume. Patients who received analgesic/sedative treatment (n=17) consisting of ketamine + benzodiazepine alternated with opioid + benzodiazepine had worse PTSD symptoms, but larger right hippocampal volume. Posttraumatic Diagnostic Scale (PDS) score and total burned surface area were significant predictors of left hippocampal volume, with smaller volume in patients who had larger burns and worse PTSD symptoms.
Schonenberg et al. (2005) found peritraumatic S-ketamine was associated with worse acute and long-term psychiatric outcomes in a retrospective study of 56 moderately injured accident victims who received ketamine (n=17), S-ketamine (n=12), or opioids (n=27) during emergency ambulance transport. Ketamine was always given with midazolam to reduce its cardiovascular effects. Of those treatments, S-ketamine exposure correlated with greater retrospectively assessed acute symptoms, including dissociation, re-experiencing, and avoidance, whereas ketamine slightly increased scores. Other relevant variables like injury severity did not differ between the groups.
One year after the incident, PTSD symptoms were more common in S-ketamine patients; they reported more re-experiencing, avoidance, and hyperarousal compared to the opioid group and greater re-experiencing and avoidance compared with ketamine. In contrast, ketamine did not differ from opioid treatment (Schonenberg et al., 2005).
A subsequent study by Shonenberg et al. (2008) supported the retrospective reports of ketamine’s acute effect after trauma discussed by Schonenberg et al. (2005). In this paper, patients were evaluated on the third day after admission for moderate accidental trauma. During emergency treatment, they were given ketamine (n=13), opioids (n=24), or non-opioid analgesics (n=13); usually ketamine was combined with midazolam. Acute Stress Disorder (ASD) symptoms were worse in the ketamine group, with elevated dissociation, re-experiencing, hyperarousal, and avoidance. Other clinical characteristics did not differ between groups.
Relevant Research (Non-Ketamine)
Lamotrigine, which reduces glutamate release, was superior to placebo in a 12-week DBRCT of 14 PTSD patients, 10 of whom received lamotrigine (Hertzberg et al., 1999). The response rate was 50% with lamotrigine compared to 25% with placebo; lamotrigine was associated with less re-experiencing and avoidance/numbing symptoms. Although this is a very small study, it suggests glutamate release inhibition could be effective in PTSD even though that mechanism appears to be in opposition to how ketamine works.
The GluN2B-NMDAR antagonist ifenprodil was effective in three females with long-term PTSD associated with early life sexual abuse (Kishimoto et al., 2012). At 60 mg/d (oral), it was highly effective after 2-4 weeks. These patients had previously failed to experience a reduction in flashbacks with standard antidepressants, although those medications did reduce depressive symptoms. In one patient who responded to ifenprodil, flashbacks increased when treatment was stopped for a few days, and they subsided again upon restarting. Ifenprodil was well-tolerated.
5.2 Obsessive-Compulsive Disorder (OCD)
Supporting Research
Rodriguez and colleagues (2011) compared ketamine (0.5 mg/kg IV, 40-min) with placebo in a female with treatment-resistant OCD. She had severe OCD symptoms (Y-BOCS = 30) and minimal depressive symptoms. Ketamine quickly produced full resolution of her obsessions, with a partial re-occurrence shortly after the end of the infusion and a plateau in efficacy until Day 7, at which time her obsessions were back to baseline. It was well-tolerated, only producing transient lightheadedness, dry mouth, and feelings of unreality.
Ketamine had persistent efficacy in a DBRCT crossover study of 15 unmedicated OCD patients with near-constant obsessions (>8 h/day) (Rodriguez et al., 2013). They were treated with a single infusion (0.5 mg/kg IV, 40-min) compared with placebo; their baseline OCD symptoms were severe (Y-BOCS = 28.2) and 2/15 had mild-to-moderate comorbid depression. The infusions were separated by at least one week, but there was a significant carryover effect post-ketamine such that they did not return to baseline by the second treatment. To eliminate the influence of carryover effects, the authors analyzed the first phase of treatment in which eight people received ketamine and seven received placebo.
There was a large effect (d>0.8) on OCD scores at mid-infusion, 230 min, and Day 7; 50% of ketamine patients responded on Day 7 compared with 0% of placebo patients (Rodriguez et al., 2013). Ketamine mildly increased BP and HR in all patients; aside from those effects, the most common side effects were dissociative symptoms like feelings of unreality (n=14) and distortions of time (n=13). Positive psychotic symptoms were reported, particularly unusual thought content (n=12) and conceptual disorganization (n=10). There were no side effects in the week after the infusion. Ketamine, norketamine, and DHNK concentrations did not correlate with response or acute psychological side effects.
In an open-label trial of 10 unmedicated OCD patients with near-constant intrusive obsessions, one ketamine infusion (0.5 mg/kg IV, 40-min) alleviated OCD when followed by 10 exposure therapy sessions over a two-week period (Rodriguez et al., 2016). 9/10 completed the treatment series, eight of whom had a rapid reduction in obsession severity with ketamine, which persisted to at least 230 min in seven. At the end of Week 2, 63% met the response criterion of a ≥35% reduction in Y-BOCS. From baseline to four weeks post-infusion, Y-BOCS was also significantly lower. One patient did not benefit, while another had sustained remission for at least six months.
Opposing Research
Bloch et al. (2012) found no effect of ketamine (0.5 mg/kg IV, 40-min) in an open-label study of 10 patients with severe treatment-resistant OCD; seven had comorbid depression and seven were on psychiatric medications (SSRI=7; antipsychotic=3; NAC/riluzole=1). No patient had a ≥35% reduction in OCD symptoms during the first three days, yet 4/7 patients with comorbid depression had an antidepressant response. Ketamine was also ineffective when specifically looking at obsessional versus compulsive symptoms. Two of the non-depressed patients had dysphoria, anxiety, and passive suicidal ideation within the first two days after ketamine.
5.3 Anxiety Disorders
Supporting Research
Ketamine administered in an escalating manner (0.25, 0.5, and 1 mg/kg SC) over the course of three weekly treatments alleviated anxiety in treatment-resistant anxiety disorder patients who did not have comorbid depression (Glue et al., 2017). Patients could continue their existing pharmacotherapy and psychotherapy; at baseline, they had severe anxiety (HAM-A = 26), specifically GAD (83%) and SAD (75%). Anxiety was reduced within one hour and the anxiolytic effect lasted 3-7 days.
Dose-dependence was seen for the anxiolytic, dissociative, and hemodynamic effects (Glue et al., 2017). 10/12 responded at 0.5-1 mg/kg, whereas 0.25 mg/kg caused relatively minor effects. Every patient had dissociative effects beginning 5 min post-injection and peaking at 20-30 min, returning to baseline by 60 min. Two patients said the experience was very intense and they felt out of control with the 1 mg/kg dose. At 1 mg/kg, the average increases in hemodynamic parameters were: +9.4 mmHg for SBP, +7.5 mmHg for DBP, and +4.5 bpm for HR.
Glue et al. (2018) studied the long-term outcome of initial ketamine responders who elected to receive three months of maintenance ketamine. Of the 21 responders, one declined maintenance therapy and two dropped out during maintenance (Weeks 4 and 9), leaving 18 patients for the full assessment. 15 patients had GAD and 18 had SAD; additionally, four had panic disorder and the mean HAM-A at entry was 12.6 (down from 25 before the first ketamine injection). Every patient decided to remain on 1 mg/kg (SC). Four patients received twice weekly injections because of faster anxiety re-occurrence, while the other 16 received weekly injections.
There was a progressive reduction in HAM-A and Fear Questionnaire (FQ) ratings until 3.5 and 7.5 weeks, respectively (Glue et al., 2018). Social functioning was improved in 16 patients and work functioning improved in 11. Patients often reported being more functional in meetings and presentations, and they could attend social events (e.g. parties). During three months of post-treatment follow-up, five remained well, eight had a partial re-emergence of symptoms, and five had a complete re-emergence within two weeks of the final dose.
Tolerance to the acute dissociative effects was reported, with a mean CADSS score of 20 at Week 1 and 8.8 at Week 14; more specifically, six patients had a decline in dissociation, three experienced consistently large effects, and one had a gradual increase in dissociation towards the end of the study (Glue et al., 2018). The average increase in SBP and DBP was ~10 mmHg throughout the study, with no evidence of tolerance. One significant adverse event occurred: a patient had substantially elevated BP (207/103 mmHg) and delirium within five minutes of the injection, which resolved within 15 min—because of the fast onset, the authors suspected ketamine was accidentally injected intravenously. No patient reported memory impairment or urological problems (e.g. symptoms of cystitis).
Ketamine was effective in a DBRCT of 18 adults with SAD who received one infusion (0.5 mg/kg IV, 40-min) (Taylor et al., 2018). They could continue with stable pharmacotherapy during the study (5 were on SSRIs, 2 were on benzodiazepines); response was considered a ≥35% LSAS reduction or ≥50% VAS-Anxiety reduction. LSAS response was more likely with ketamine (33% vs. 0%), as was VAS-Anxiety response (89% vs. 53%). Patients had a significant LSAS reduction post-ketamine on Days 2, 5, and 10; ketamine was also superior on STAI-S ratings on Days 1, 7, and 10. Acute dissociation (CADSS) did not correlate with LSAS response. Depressive symptoms did not significantly improve in the 12 patients with baseline comorbid depression.
Anxiety was reduced in a double-blind study comparing different doses of ketamine (0.25, 0.5, 1 mg/kg SC) with midazolam (0.01 mg/kg SC) in 12 patients with treatment-resistant GAD (n=10) and/or SAD (n=12); comorbid depression was not permitted (Shadli et al., 2018). The ascending doses were given at one-week intervals and anxiety was assessed using the FQ and HAM-A. There was a dose-dependent improvement in FQ, but not HAM-A, at 120 min, with an apparent ceiling effect at 0.5 mg/kg:
- FQ (mean reduction from baseline): 21% with midazolam; 31% with 0.25 mg/kg; 43% with 0.5 mg/kg; and 42% with 1 mg/kg
- HAM-A (mean reduction from baseline): 46% with midazolam; 42% with 0.25 mg/kg; 70% with 0.5 mg/kg; and 67% with 1 mg/kg
Compared to baseline, ketamine reduced EEG power in the δ frequency and increased γ power two hours after administration (Shadli et al., 2018). However, the only predictor of ketamine’s effect on FQ scores was reduced theta frequency.
5.4 Substance Use Disorders
Supporting Studies
Ketamine increased motivation to quit and reduced cue-induced craving in eight cocaine-dependent people studied in a DBRCT crossover trial of ketamine (0.41 mg/kg and 0.71 mg/kg IV, 50-min) versus lorazepam (Dakwar et al., 2014). The subjects were not seeking treatment or abstinence when they entered the study; the infusions were separated by 48 h during a 9-day hospital stay in which the subjects were abstinent for three days before the first infusion. Compared with lorazepam, ketamine (0.41 mg/kg) increased scores on the URICA (University of Rhode Island Change Assessment) by 60% over baseline and cue-induced craving at 24 h was reduced to a similar degree. Increasing the dose to 0.71 mg/kg seemed to further reduce cue-induced craving, but those results may have been impacted by persistent effects from the smaller preceding dose.
During a 4-week follow-up period, no subject voluntarily enrolled in substance use disorder treatment, but they reported a significant reduction in cocaine use (Dakwar et al., 2014). The average amount spent on cocaine fell from $149/day to $11/day and the number of days with use declined from 22 days per 28-day period to 5 days per 28-day period. Subjects who remained abstinent for at least two weeks (n=4), which was confirmed using urine toxicology, had higher post-ketamine URICA scores, suggesting ketamine can trigger a meaningful desire to change one’s use without a preexisting intent to pursue abstinence or treatment.
Assessing the role of spiritual experiences in the same group of cocaine-dependent individuals, Dakwar et al. (2014) reported greater mystical-type effects shortly after ketamine administration compared with lorazepam and the 0.71 mg/kg dose was more active than 0.41 mg/kg. Ketamine’s mystical-type effects, but not its dissociative effects, correlated with increased motivation to quit cocaine one day later; no correlation with altered cue-induced craving was found.
Section 6: Biomarkers, Predictors, and Correlates
6.1 Brain-Derived Neurotrophic Factor (BDNF)
Normal BDNF function seems to be required for the antidepressant-like effects of ketamine in animals and BDNF has for many years been associated with mood disorder. One way BDNF function can be impaired in humans is with the reduced function Met allele of the BDNF gene, which has been associated (often controversially) with stress susceptibility and psychiatric conditions like depression. BDNF activity appears to be reduced by the Val/Met and Met/Met genotypes compared to Val/Val. The prevalence of this polymorphism varies between ethnic groups.
The Met allele of the BDNF gene was found in 41% of Japanese people (n=151) compared with 18% of Americans (n=133) and the Val/Val (normal function) genotype was present in 34% of Japanese, 49% of Italians, and 68% of Americans (Shimizu et al., 2004).
Supporting Research
Among 62 MDD patients treated with ketamine (0.5 mg/kg IV, 40-min) in four studies, 41 had the Val/Val BDNF genotype, 19 had one Met substitution, and two had a Met/Met genotype; those with the Val/Val genotype were more likely to have an antidepressant response to ketamine at 230 min (Laje et al., 2012). The mean HDRS reduction was 24% in Met carriers and 41% in Val/Val patients. Most patients had European ancestry (n=58), while four self-reported African ancestry.
Baseline serum neurotrophin levels in treatment-resistant BD inpatients (n=25) did not differ between people who responded to ketamine (0.5 mg/kg IV, 40-min) and those who did not, but the nonresponder group had a reduction in BDNF on Days 7 and 14; they also had a trend towards lower NGF on Day 7 and a significant reduction on Day 14 (Rybakowski et al., 2013). These patients were receiving mood stabilizers during the study. Other neurotrophins (NT-3, NT-4, and GDNF) did not differ between groups. Responders had a quantitative nonsignificant increase in BDNF on Days 7 and 14. The response and remission rates over time were: 6 h (4% and 0%), 24 h (24% and 16%), Day 7 (52% and 32%), and Day 15 (52% and 48%).
Haile et al. (2014) treated TRD patients in a DBRCT of ketamine (n=15) versus midazolam (n=7) and observed higher plasma BDNF in ketamine responders at 240 min; BDNF did not differ between midazolam responders and nonresponders. This study was part of the trial published by Murrough et al. (2013). The response rate at Day 7 was 47% (7/15) with ketamine and 29% (2/7) with midazolam. In the ketamine group, there was a negative correlation between MADRS score and BDNF level, with plasma BDNF at 240 min predicting MADRS score at 240 min, 24 h, 48 h, and 72 h, but not at Day 7.
A trial of repeated ECT versus ketamine in TRD patients (n=35) reported lower serum BDNF in patients compared to healthy controls (n=20); there was no baseline difference between the ECT and ketamine groups (Allen et al., 2015). Patients received twice-weekly bitemporal ECT (n=17) or up to three ketamine infusions (n=18; 0.5 mg/kg IV, 40-min) while continuing with their existing medications; the ECT group received a median of eight sessions. Both treatments reduced depression severity and most ketamine-treated patients responded at 2 h. Those who responded to ketamine one week after treatment had higher BDNF at that time, although their levels had not normalized to control levels. Interestingly, BDNF was not elevated 2 h after ketamine, indicating that effect develops over time. ECT did not affect BDNF in nonresponders or responders.
Opposing Research
In Taiwanese MDD patients (n=71) treated with ketamine (0.2 or 0.5 mg/kg IV, 40-min) in a DBRCT, BDNF genotype did not affect response (Su et al., 2017). The Val/Val genotype was present in 17% (n=12), Val/Met was in 56% (n=40), and Met/Met was in (27%). Met carriers had a 34% response rate compared with a 25% response rate among Val/Val patients.
6.2 Dissociative and Psychotomimetic Effects
Supporting Research
Acute psychotomimetic effects correlated with ketamine-induced depression alleviation after a single dose (0.27 mg/kg over 10-min + 0.27 mg/kg over 20-min) in 27 hospitalized depressed patients studied in a DBRCT crossover trial (Sos et al., 2013). Patients were maintained on a stable antidepressant during the study. BPRS total score weakly correlated with antidepressant efficacy on Day 7 and there was a trend-level correlation on Days 1 and 4; ketamine was superior to placebo at all timepoints. The response rate was higher with ketamine than placebo on Day 1 (37% vs. 4%), Day 4 (41% vs. 4%), and Day 7 (37% vs. 11%). Ketamine did not cause serious adverse effects and it only transiently produced psychotomimetic effects; among its typical side effects were confusion, dissociation, perceptual disturbance, mild BP increase, emotional blunting, and euphoria.
Luckenbaugh et al. (2014) analyzed data from 108 treatment-resistant depression patients (MDD=74; BD=34) who were treated with ketamine (0.5 mg/kg IV, 40-min) in two double-blind and one open-label study; patients were unmedicated except for BD patients maintained on lithium or valproate. Dissociative effects at 40 min correlated with antidepressant effects at 230 min and Day 7, but not Day 1. In contrast, psychotomimetic effects (YMRS, BPRS) did not correlate with depression reduction.
An analysis of data from 126 treatment-resistant patients (MDD=84; BD=42), including the 108 patients assessed in the report from Luckenbaugh et al. (2014), reported that greater dissociative effects (CADSS) only significantly predicted greater HDRS depression reduction on Day 7 in 1/3 studies; across the studies there was a negative, nonsignificant correlation between CADSS score and HDRS score at 230 min (Niciu et al., 2018). The depersonalization subscale of CADSS was more consistently associated with antidepressant effects than derealization, with greater depersonalization predicting greater treatment response across the study. Interestingly, the study that reported the strongest correlation involved unmedicated patients, unlike the other two studies, and they had a more severe course of illness.
Opposing Research
Dissociative effects were not predictive of depression improvement in a study of 10 MDD patients treated with ketamine (0.5 mg/kg IV, 40-min) compared with placebo (Valentine et al., 2011).
6.3 Family History of Alcohol Dependence
One correlation that has repeatedly appeared in the ketamine literature is that superior effects occur in patients with a family history of alcohol dependence, usually in a first-degree relative or multiple second-degree relatives. Alcohol is known to act on NMDARs and healthy people with a family history of alcohol dependence seem to experience less perceptual disturbance and dysphoria from ketamine, but the explanation for this putative connection in depression treatment is unknown.
Supporting Research
Phelps and colleagues (2009) observed greater acute depression reduction in an open-label study of 26 TRD patients treated with ketamine (0.5 mg/kg IV, 40-min) in those with a family history of alcohol dependence. Family history information was available for 23 patients, 12 (53%) of whom had a positive family history (FHP). Lower MADRS score in FHP patients was seen at 120 min, with trends at 80 min and 230 min. At 230 min, the response rate was 67% among FHP patients compared to 18% for patients without a positive history. And the difference in remission (42% vs. 9%) approached significance. The same results were found using the HDRS and BDI at 80 min through 230 min. FHP subjects also had less dysphoria (BPRS-dysphoria) at 120 min and 230 min, but other psychotomimetic effects were similar.
Family history of alcohol dependence correlated with superior depression reduction (MADRS) in two DBRCT crossover trials of 33 treatment-resistant BD patients who were treated with ketamine (0.5 mg/kg IV, 40-min) while receiving lithium or valproate (Luckenbaugh et al., 2012). 36% (12/33) had a positive history; that group also had a longer illness duration, a more substantial lifetime history of substance abuse or dependence, and more valproate use. Subjects with a positive family history experienced less acute psychotomimetic and dissociative effects.
A significant correlation between antidepressant response and family history of alcohol dependence was reported in an analysis of data from four studies of 108 patients who received ketamine (0.5 mg/kg IV, 40-min) for treatment-resistant MDD (n=74) or BD (n=54). Patients were unmedicated in two studies ad 3/4 trials were DBRCTs, while one was an open-label study. The correlation was significant on Days 1 and 7, but not at 230 min. Higher BMI also correlated with greater depression reduction at 230 min and Day 1, and no history of suicide attempt correlated with greater improvement on Day 7.
6.4 Cognitive Functioning
Supporting Research
Lower baseline attention correlated with a greater likelihood of response among TRD patients (n=15) treated with six infusions (0.5 mg/kg IV, 40-min) over a 12-day period (Shiroma et al., 2014). Murrough et al. (2013) also reported a higher likelihood of response to a single infusion (0.5 mg/kg IV, 40-min) in people with slower processing speed at baseline and among older subjects in an open-label trial of 25 unmedicated TRD patients. Responders at 24 h (n=16) had more baseline cognitive impairments, particularly in tasks involving working memory and processing speed.
6.5 Response Characteristics
Pennybaker and colleagues (2017) reviewed four studies of 122 unmedicated MDD or BD patients who received ketamine (0.5 mg/kg IV, 40-min). Response on Day 1 was predictive of response at two weeks; in particular, levels of sadness were an independent predictor of Week 2 outcome. 76% of patients followed their study protocol for at least two weeks after infusion; therefore, the analysis was based on 93 patients. The mean MADRS change on Day 1 was -34%, which declined to -17% at two weeks, yielding a response rate of 33% on Day 1 and 13% by Week 2.
Improved sadness (apparent and reported), inability to feel, and difficulty concentrating on Day 1 were most strongly correlated with outcome at two weeks (Pennybaker et al., 2017). Dissociation at 40 min and a positive family history of alcohol use disorder in a first-degree relative also correlated with efficacy.
6.6 Brain Imaging
Supporting Research
Increased ACC activation in response to fearful faces at baseline positively correlated with response to a single infusion (0.5 mg/kg IV, 40-min) in a study of 11 unmedicated MDD patients compared with 11 healthy controls (Salvadore et al., 2009). Although 73% of patients had comorbid anxiety, the correlation between antidepressant effects and increased baseline ACC activation was minimally affected by controlling change in anxiety. Repeated exposure to fearful faces reduced ACC activation in healthy controls, but the opposite happened at baseline in MDD patients. Both groups had a decrease in right amygdala activity after repeated exposure to fearful faces, but the change in activity among patients negatively correlated with antidepressant response such that larger decreases were associated with superior response at 230 min. There was a significant reduction in MADRS score from 32 at baseline to 20 at 230 min; anxiety was also reduced, with the mean HAM-A score falling from 23 to 14.
Ketamine treatment was associated with reduced metabolism in the habenula, insula, ventrolateral PFC (vlPFC), and dlPFC of the right hemisphere, whereas metabolism increased in the bilateral occipital, right sensorimotor, left parahippocampal, and left inferior parietal cortices, as seen at 2 h using FDG-PET (Carlson et al., 2013). Those findings were reported in an open-label study of 0.5 mg/kg (IV, 40-min) given to unmedicated TRD patients (n=20). The mean reduction in MADRS score was 30% at 230 min and 30% (6/20) of patients had a response. Depression reduction positively correlated with altered metabolism in the right superior and middle temporal gyri (STG/MTG) and cerebellum, whereas depression reduction inversely correlated with changes in the right parahippocampal gyrus and temporoparietal cortex. The strongest correlations were with metabolic changes in the STG/MTG regions.
FDG-PET scan data from 21 BD patients maintained on lithium or valproate in two DBRCT crossover trials showed that depression reduction at 230 min correlated with metabolic increases in the right ventral striatum (Nugent et al., 2014). Nine patients responded to ketamine at 230 min, with a mean post-infusion MADRS score of 29 with placebo and 18 with ketamine. Ketamine was associated with reduced glucose metabolism in the left hippocampus, lingual gyrus, and right parahippocampal gyrus. Patients with the highest glucose metabolic rate in the sgACC during the placebo infusion had the greatest depression reduction post-ketamine. There was a trend-level inverse correlation between metabolic rate in the left hippocampus and MADRS score, with lower metabolism correlating with worse symptoms.
Patients who had a smaller left hippocampus had a larger response to ketamine (0.5 mg/kg IV, 40-min) at 24 h in a DBRCT comparing ketamine (n=16) with midazolam (n=8) in unmedicated TRD patients (Abdallah et al., 2015). MRI was used to assess brain region volume; successful scans were available for 13 ketamine-treated patients and six midazolam patients.
Seven hours after an open-label infusion of ketamine (0.5 mg/kg IV, 40-min) in MDD patients (n=13), connectivity between the amygdala and an insulo-temporal region was reduced, approaching the level of connectivity observed in healthy controls (n=18) not given ketamine (Nugent et al., 2016). Magnetoencephalography (MEG) was used to study these changes. MDD patients had a reduction in connectivity to limbic regions post-ketamine and that reduction was present regardless of baseline connectivity.
By splitting unmedicated MDD patients into those with low baseline mPFC GABA (n=13; ‘glutamate-based depression’ (GBD)) and high GABA (n=13), as determined using 1H-MRS, Abdallah et al. (2017) found bilateral NAc volume at baseline (using MRI) was larger in non-GBD depression compared with healthy controls (n=26), but there was no difference in GBD patients. Right NAc volume correlated with mPFC Glx level; that correlation was not seen in the left NAc. Left NAc volume was larger in the full patient sample than in control subjects (599 vs. 489 mm3), without a difference in the right NAc.
In a separate cohort of unmedicated TRD patients (n=16) treated with ketamine (0.5 mg/kg IV, 40-min), treatment was associated with a small reduction in left NAc volume at 24 h; the effect was specifically reported in remitters, not non-remitters (Abdallah et al., 2017). 62% (10/16) had a response at 24 h and 38% (6/16) remitted. Ketamine exposure was also associated with a trend towards increased left hippocampal volume among remitters. Grouping patients by baseline hippocampal volume showed that ketamine reduced left and right NAc volume in those with larger left hippocampal volume, but no significant change occurred in patients with smaller total or left hippocampal volume.
Opposing Research
Ketamine caused remission in 10/11 (91%) MDD patients at 230 min and an increase in mPFC Glx (glutamate+glutamine) and GABA cycling, with a peak increase of ~40% around 26 min; those effects were measured using 1H-MRS (Milak et al., 2016). At 24 h, 9/11 patients met remission criteria and 7/10 were still remitted on Day 3. Although ketamine affected prefrontal Glx and GABA, those changes did not correlate with efficacy. Psychotic symptoms (BPRS total) were reduced from baseline to 230 min, specifically with stable positive symptoms and reduced scores on the BPRS subscales for anxiety-depression and anergia (lack of energy). Ketamine caused no or mild adverse cognitive and dissociative effects acutely. Change in Glx and GABA did not correlate with BPRS score.
6.7 Kynurenine and Arginine
Supporting Research
Ketamine reduced plasma kynurenine and increased arginine at 4 h when administered at 0.5 mg/kg (IV, 40-min) to unmedicated TRD patients (n=29) compared with healthy controls (n=25) in a DBRCT crossover study (Moaddel et al., 2018). Responders had a larger reduction in kynurenine and kynurenine/tryptophan ratio at 4 h and a larger increase in circulating arginine, whereas TRD patients did not differ from controls at baseline for either metabolite. There was an increase in circulating sphingomyelins post-ketamine, but they did not correlate with response.
6.8 Serine
Supporting Research
Moaddel et al. (2015) found lower plasma D-serine and L-serine levels at baseline in unmedicated MDD patients who responded to ketamine (0.5 mg/kg IV, 40-min) at 230 min (n=8) versus nonresponders (n=13). Baseline D-serine inversely correlated with dissociative symptoms (CADSS) at 40 min and CADSS score was higher in responders. When baseline D-serine and BMI were included in a linear regression model, D-serine was a significant predictor of response, but BMI was not; baseline D-serine accounted for 60% of the variance in response. In contrast, in a model that considered BMI and baseline L-serine, both were predictors of response; L-serine accounted for 69% of variance, which increased to 75% when considering both.
Ketamine also reduced plasma D-serine by 26% at the end of the infusion in responders and by 19% in nonresponders (Moaddel et al., 2015). D-serine continued to decline for a week after the infusion, with a 39% reduction in responders and a 28% reduction in nonresponders on Day 7. Responders had lower levels than nonresponders at 120 min, 230 min, and Day 1. Ketamine did not affect L-serine. At baseline, nonresponders had an average D-serine concentration of 4.68 μM, which was higher than the 3.02 μM concentration in responders.
Relevant Research (Non-Ketamine)
A study of TRD patients (n=27) compared with non-TRD patients (n=8) and controls (n=15) reported that people who did not respond to five weeks of antidepressant treatment had lower serum aspartate, asparagine, serine, threonine, and taurine (Maes et al., 1998). Five weeks of treatment reduced serum aspartate, glutamate, and taurine, while increasing glutamine. At baseline, there was no difference between the groups. This in contrast with ketamine, where response was associated with lower serine (Moaddel et al., 2015).
Section 7: Antidepressant Mechanism
7.1 Glutamatergic Activity
Introduction to NMDARs
N-methyl-D-aspartate receptors (NMDARs) are protein complexes consisting of subunits that associate with each other to form an ion pore, producing an ionotropic receptor. They facilitate the entry of cations—particularly sodium (Na+), potassium (K+), and calcium (Ca2+)—into the cell. An NMDAR has four subunits, making it a tetramer, and more precisely a heterotetramer because those subunits are not identical. Every receptor complex has two obligatory GluN1 subunits that must be present for a functional receptor to exist. Aside from GluN1, receptors often contain two GluN2 subunits, but some contain GluN3 subunits in place of one or both GluN2. GluN2 subunits are encoded by four genes, yielding variations of the protein referred to as GluN2A-D; those variations can affect the receptor’s properties (Vyklicky et al., 2014).
The prototypical GluN1/N2 NMDAR is activated when two agonist molecules (usually glutamate) and two coagonist molecules (often glycine) are bound. Regardless of neuronal activity, glycine is bound to many receptors because its basal CSF concentration is 4.2 μM, much higher than its EC50 of ~1 μM (Vyklicky et al., 2014). Different molecules can behave as coagonists, like D-serine and D-alanine, while weaker coagonists include L-alanine and L-serine. With its low EC50 of 0.65 μM, D-serine is physiologically important and it may be the primary coagonist at synaptic NMDARs, with glycine predominating at extrasynaptic receptors. Aside from L-glutamate, other molecules can function as agonists, including NMDA, D-aspartate, and L-aspartate.
A tightly regulated glutamate release and uptake system is present at the “tripartite synapse,” which refers to the presynaptic glutamate-releasing neuron, the postsynaptic glutamate-binding neuron, and the surrounding glial cells that express glutamate transporters to remove glutamate from the synapse. Proper functioning of glutamate transporters is vital for preventing overactivation of glutamate receptors; they are responsible for reducing the synaptic glutamate concentration from ~1 mM immediately after presynaptic release to only a nanomolar concentration at rest, thereby minimizing ‘at rest’ glutamate receptor activation (Vyklicky et al., 2014).
Spontaneous Receptor Activity
Spontaneous subthreshold activity was observed at motor nerve endings in muscle fibers in the 1950s (Fatt and Katz, 1952) and it is now understood the underlying mechanism for that kind of activity involves presynaptic glutamate release at rest, which produces miniature excitatory postsynaptic currents (mEPSCs) (Sutton et al., 2004). This activity is associated with postsynaptic protein synthesis regulation, particularly a reduction in protein synthesis in the case of NMDAR-mediated mEPSCs.
Activity-dependent glutamate release promotes protein synthesis; therefore, the in vitro application of tetrodotoxin (TTX), which attenuates action potential (AP)-induced neurotransmitter release but does not prevent mEPSCs, reduces protein synthesis (Sutton et al., 2004). However, if mEPSCs are also blocked by administering TTX alongside AMPAR and NMDAR antagonists, there is an increase in dendritic protein synthesis. Protein synthesis was increased just with coadministration of TTX and the NMDAR antagonist D-APV, but the effect was greater when AMPARs were also blocked using CNQX.
When NMDARs are blocked in cultured hippocampal neurons during action potential inhibition by TTX, the amplitude of AMPAR-mEPSCs is increased (Sutton et al., 2006). The increase in AMPAR-mediated currents is dependent on a local increase in dendritic protein synthesis and membrane insertion of synaptic AMPARs, which initially involves an increase in synaptic GluR1 (AMPAR subunit) expression and more GluR2-lacking AMPARs at synaptic sites; GluR2-lacking AMPARs are replaced by GluR2-containing receptors over time. This suggests synaptic plasticity is regulated by the tonic suppression of dendritic protein synthesis by NMDAR-mEPSCs.
The mechanism underlying protein synthesis regulation by mEPSCs involves eukaryotic elongation factor 2 (eEF2). Sutton et al. (2007) demonstrated that spontaneous activity strongly promotes the inactivation of eEF2 via increased phosphorylation, whereas phasic AP-mediated activity maintains eEF2 in a more dephosphorylated (active) state. When eEF2 kinase (eEF2K) was inhibited by rottlerin or NH125, protein synthesis increased, consistent with a role for eEF2 in the translation suppression caused by NMDAR-mEPSCs, which appear to activate eEF2K.
Ketamine activates eEF2 and induces synaptic plasticity, effects that were mimicked by depleting the pool of neurotransmitter vesicles involved in spontaneous neurotransmission using folimycin, an ATPase blocker that inhibits the reacidification of vesicles, preventing them from refilling with more neurotransmitter after endocytosis (Nosyreva et al., 2013). Folimycin reduced mEPSCs and eEF2 phosphorylation to the same extent as ketamine when applied to hippocampal slices.
Extrasynaptic NMDARs
In addition to synaptic NMDARs, there are postsynaptic receptors on spine necks, dendritic shafts, and somas, which are located away from the synapse (Zhou et al., 2014). Extrasynaptic NMDARs can contribute to tonic currents triggered by ambient glutamate originating from spillover of synaptic glutamate, particularly when synaptic glutamate is abnormally elevated, like in brain trauma. While they are often associated with neurodegenerative signaling, extrasynaptic NMDARs can contribute to synaptogenesis, neuritogenesis (i.e. axon or dendrite formation), and the migration and differentiation of neurons. In hippocampal neurons, they are located at various distances from synapses and are localized along dendrites and spines, usually in close contact with axons/terminals or glia (Petralia et al., 2010).
The Role of NMDARs
The most prominent effect of ketamine is NMDAR antagonism (Anis et al., 1983). It is an open-channel, voltage-dependent, noncompetitive antagonist that inhibits the influx of cations when it occupies the channel. Although this property certainly exists, there is disagreement as to what role NMDAR antagonism directly plays in the mechanism of action for its antidepressant effects. A few observations have complicated theories that emphasize the importance of NMDAR antagonism.
Ketamine’s anesthetic and analgesic effects have long been associated with NMDAR antagonism, so when new effects (e.g antidepressant potential) were observed, naturally they were associated with NMDAR antagonism. Yet interest in NMDAR antagonism as a novel target for antidepressants produced studies on other drugs with that property, including investigational drugs and existing medications like memantine, most of which have failed to replicate the effects of ketamine in humans (Zarate et al., 2013; Lenze et al., 2012; Smith et al., 2013). Furthermore, if NMDAR antagonism is the most important action, S-ketamine (the more potent antagonist) would be expected to be a more potent antidepressant, yet in numerous animal studies R-ketamine is superior to S-ketamine.
This either suggests that ketamine has unique properties at NMDARs that distinguish it from other antagonists or that other mechanisms are at least contributing to the effects. Regardless of its initial effect on NMDARs or other targets, the antidepressant effects seem to come from downstream changes in neurotrophin signaling and synaptic plasticity, but there are multiple ways to trigger those downstream effects.
Disinhibition Hypothesis
At least two hypotheses prioritize an initial role of NMDAR antagonism in the antidepressant effects of ketamine: the disinhibition and direct hypotheses. The disinhibition hypothesis asserts that ketamine preferentially inhibits GABAergic inhibitory interneurons (iINs) by blocking NMDARs, which then disinhibits neurons in regions like the PFC and hippocampus because they receive less inhibitory input. Associated with that excitatory effect is a prefrontal increase in glutamate, which was demonstrated by Moghaddam and colleagues in 1997 through their research on subanesthetic ketamine in rats (Moghaddam et al., 1997). In contrast, prefrontal extracellular glutamate is reduced by larger anesthetic doses. Through its excitatory effect on pyramidal neurons, ketamine could increase protein synthesis and synaptic plasticity.
One of the major targets that would be affected by elevated extracellular glutamate is AMPAR, another type of glutamate receptor. Sure enough, AMPAR antagonism attenuates the antidepressant effects of ketamine and many of its associated biochemical effects (Maeng et al., 2008; Koike et al., 2011).
The hypothesis that ketamine preferentially affects iINs remains a topic of debate. One idea related to this hypothesis is that NMDARs on iINs are tonically active to maintain inhibitory synaptic tone (Miller et al., 2016), but it has yet to be firmly established that those interneurons—such as fast-spiking (FS) parvalbumin (PV)-expressing interneurons (e.g. basket cells and chandelier cells) in the cortex and hippocampus—have properties that make them uniquely important. There are many other cells that express NMDARs, although some studies have supported ketamine’s preference for iINs in vitro and in rodents. Its proposed selectivity is largely attributed to iINs being more tonically active than excitatory pyramidal neurons such that a larger proportion of their NMDARs are not blocked by magnesium and can therefore bind ketamine (Duman, 2018).
Supporting this hypothesis, Neske et al. (2015) found PV-expressing iINs were the most active cell type among: mouse pyramidal neurons and interneurons expressing PV, somatostatin (SOM), vasoactive intestinal peptide (PIP), or neuropeptide Y. Cultured cortical PV interneurons differ from pyramidal neurons in that they have a five-fold higher GluN2A/GluN2B ratio (Kinney et al., 2006). Ketamine selectively reduced PV and GAD67 immunoreactivity in those interneurons and its effects were mimicked by the GluN2A-selective antagonist NVP-AAM077, but only partially mimicked by the GluN2B-selective antagonist Ro 25,6981. NMDAR antagonist-induced reduction in PV and GAD67 immunoreactivity was attenuated when neurons were coexposed to the calcium channel opener BayK or the group 1 metabotropic glutamate receptor agonist BHPG. Subunit composition differences may not substantially contribute to ketamine’s selectivity since it has a similar affinity for GluN2A- and GluN2A-containing receptors (Kotermanski and Johnson, 2009).
PV-expressing interneurons have been of interest as a possible target for NMDAR antagonists both because of their relevance to depression physiology and because PV interneurons have been implicated in psychotic disorders, which NMDAR antagonists are considered a model for (Xu et al., 2015; Lewis and Moghaddam, 2006; Schobel et al., 2013). Additionally, NMDAR antagonists cause a loss of PV interneurons (Yang et al., 2016) and altered activity of those neurons correlates with schizophrenia (Beasley and Reynolds, 1997; Gonzalez-Burgos et al., 2015), possibly contributing to the cognitive deficits observed in that disorder (Lewis and Moghaddam, 2006).
Hippocampal slices treated with ketamine showed a reduction in inhibitory synaptic input onto pyramidal cells in the CA1 region at an antidepressant-relevant concentration of 1 μM (Widman and McMahon, 2018). This increased the probability of action potential generation within postsynaptic neurons without altering their intrinsic excitability, i.e. it did not increase the number of APs generated by direct current injection. Because the disinhibition hypothesis suggests there is normally a fair amount of spontaneous tonic inhibitory input onto those cells, the researchers tested whether ketamine reduces spontaneous inhibitory postsynaptic current (sIPSC) frequency in pyramidal cells, which it did. In case ketamine-induced inhibition was not actually selective for inhibitory currents, Widman and McMahon assessed its effect on spontaneous excitatory postsynaptic currents (sEPSCs) in the presence of the GABAA receptor antagonist picrotoxin, thereby blocking inhibitory currents. It failed to influence sEPSC frequency, indicating it selectively reduces sIPSC frequency via a presynaptic effect on GABAergic interneurons.
Other NMDAR antagonists were less reliable. A different study found the GluN2B-NMDAR antagonist Ro 25-6981 reduced output from hippocampal GABAergic interneurons without affecting postsynaptic pyramidal neurons (Hanson et al., 2013), but it did not cause ketamine-like effects in Widman and McMahon’s research, where it increased the strength of sIPSCs (Widman and McMahon, 2018). This could be evidence of a vital non-NMDAR effect of ketamine, a role for non-GluN2B NMDARs, or of different properties at NMDARs.
The NMDAR antagonist MK-801 mimicked ketamine by predominantly inhibiting suspected GABAergic interneurons in rats (Homayoun and Moghaddam, 2007). This inhibitory effect was followed by an increase in the firing rate of pyramidal neurons, indicating NMDAR antagonism can elevate cortical activity via disinhibition.
Ketamine preferentially inhibited putative FS interneurons in the OFC of male rats (Quirk et al., 2009). Those interneurons were associated with regulation of motivation and working memory, both of which are affected by depression. Using electrophysiological recordings, neurons in the OFC were grouped into pyramidal cells, FS interneurons, and non-FS cells based on their spike widths and firing rates. The putative FS interneurons strongly interacted with nearby pyramidal cells. Pyramidal cell firing increased just before an FS interneuron spike, then there was a decrease in pyramidal cell firing following the FS interneuron spike, i.e. there is a direct connection between these neuronal populations that results in FS interneuron firing immediately after pyramidal cell firing, thereby inhibiting the recently-active pyramidal cell.
The firing rate of putative FS interneurons was greatly reduced by ketamine (30 mg/kg SC), but the overall firing rate of pyramidal cells did not change (Quirk et al., 2009). Instead of a simple absence of effect on pyramidal cells, ketamine increased firing rate variability, with some cells showing large increases and others showing a reduction in firing, suggesting there may be two pyramidal neuron populations in the rat OFC; one population is more active post-ketamine and the other is less active.
Potentially Opposing Research
In the prefrontal region of juvenile rats, fast-spiking interneurons exhibit substantial NMDAR- and AMPAR-mediated currents, but the NMDAR-mediated currents decline in importance during cortical development (Wang and Gao, 2009). By adulthood, 74% of FS interneurons did not have NMDAR-mediated currents, suggesting this group of PFC interneurons may not be a primary target for ketamine if NMDARs already exert relatively little influence over their activity.
If it is vitally important to reduce inhibitory activity from GABAergic interneurons, perhaps reducing GABAergic activity through other means could mimic ketamine, yet studies are mixed on this point as well. The GABAA antagonist picrotoxin did not affect FST behavior in mice nor did it increase BDNF synthesis (Autry et al., 2011). In contrast, Slattery and colleagues (2011) found that reducing activity in the infralimbic PFC with the GABAA agonist muscimol caused antidepressant-like effects in the FST, whereas activating that region with the GABAA antagonist bicuculline was ineffective.
The acute (30 min) antidepressant-like effects of ketamine in female mice were enhanced by GABAA agonism (Rosa et al., 2016). A subeffective dose of muscimol combined with subeffective ketamine (0.1 mg/kg IP) reduced TST immobility at 30 min, while the opposite was seen with the GABAB agonist baclofen, which blocked the antidepressant-like effect of ketamine (1 mg/kg IP). Similarly, muscimol enhanced and baclofen blocked TST immobility reduction caused by ascorbic acid at 30 min.
Studies using genetic manipulation of receptor expression and activity have cast doubt on the role of particular interneurons. Mutant mice lacking functional NMDARs on PV interneurons—caused by genetic deletion of the obligatory GluN1 subunit—do not have antidepressant-like effects in the FST or SPT (Pozzi et al., 2014). This deletion also failed to prevent the antidepressant-like effects of ketamine at 30 min or 24 h.
Mice with disinhibited SST+ GABAergic forebrain interneurons—caused by inactivation of the GABAA γ2 subunit gene in those neurons—had an increase in inhibitory input onto CA1 pyramidal neurons and layers 2/3 of the cingulate cortex, producing anxiolytic and antidepressant-like behavior in multiple tests (Fuchs et al., 2017). Despite having an apparently opposite effect to that of ketamine, this manipulation also reduced eEF2 phosphorylation like ketamine, though with no effect on mTOR.
Supporting Research (Non-Ketamine)
Departing from research that has generated skepticism of the disinhibition theory due to the inefficacy of some GABAA antagonists, other studies have found negative allosteric modulators (NAMs) of α5-containing GABAA receptors, which are the most common subtype in the PFC and hippocampus. produce antidepressant behavioral effects. In a chronic stress model, the NAMs L-655,708 and MRK-016 were active (Fischell et al., 2015). L-655,708 restored sucrose preference in the SPT and normalized social interaction behavior in stressed animals, with an onset of efficacy within one day. It also restored the strength of excitatory transmission at stress-sensitive temporoammonic-CA1 synapses and it increased GluA1 expression. MRK-016 also restored sucrose preference and social interaction within one day, effects that remained for at least one week.
Similarly, MRK-016 reduced FST immobility at 1 and 24 h in chronically stressed mice, along with producing an anti-anhedonic response in the FUST (Zanos et al., 2017). It transiently increased EEG γ power, like ketamine, and its effects were blocked by the AMPAR antagonist NBQX. MRK-016 differed from ketamine in that it lacked impairing effects in the rotarod test, did not reduce PPI, and was not rewarding.
NMDA applied to mouse hippocampal slices produced sIPSCs mediated by GABAA receptors in CA1 pyramidal neurons and the increase in sIPSC frequency and amplitude was suppressed by TTX (Xue et al., 2011). NMDA did not affect mIPSCs. N- and P/Q-type VDCCs seem to be involved in NMDAR-mediated GABA release from interneurons since the N-type VDCC blocker ω-GVIA and the P/Q-type blocker ω-agatoxin TK partially attenuated the NMDA-induced increase in sIPSCs. The NMDAR antagonist MK-801 suppressed NMDA-induced sIPSCs, as did the nitric oxide (NO) scavenger PTIO; NMDAR activation may therefore activate an NO signaling pathway to increase GABA release.
Direct Hypothesis
According to the direct hypothesis, ketamine predominantly inhibits NMDAR signaling on excitatory neurons at rest, i.e. its effects come from the inhibition of signaling initiated by ambient and spontaneously released glutamate instead of signaling initiated by the larger glutamate concentrations produced by activity-dependent release. This view of ketamine gained traction after an influential paper was published in 2011 demonstrating at-rest NMDAR inhibition by ketamine in mice (Autry et al., 2011).
This hypothesis suggests that resting NMDAR activation by ambient glutamate deactivates intracellular eEF2, a protein that is important for protein synthesis. eEF2 is deactivated through phosphorylation via eEF2 kinase (eEF2K, aka CaMKIII), impeding protein translation. Autry et al. (2011) found that at-rest NMDAR inhibition by ketamine deactivated eEF2K, activated eEF2, and increased BDNF translation. Therefore, ketamine-induced NMDAR antagonism alleviates suppression of translation, facilitating the synthesis of important cellular proteins. eEF2K inhibitors (rottlerin and NH1255) also have rapid antidepressant effects in animals that persist for up to one week and occur alongside increased hippocampal BDNF, supporting the involvement of eEF2 in ketamine’s mechanism. However, enhanced eEF2 activity does not necessarily have to come from selectively blocking NMDARs at rest.
Ketamine’s inhibition of eEF2 phosphorylation was dose-dependent at 1, 5, and 50 μM, and those concentrations also dose-dependently reduced NMDAR-mEPSCs (Autry et al., 2011). TTX was ineffective at reducing p-eEF2, implicating spontaneous glutamate release rather than AP-induced release. Mice given ketamine or MK-801 had a reduction in hippocampal p-eEF2 within 30 min, but there was no acute change in cortical levels. As was noted by Duman (2018), it is unclear how this mechanism would apply in regions where ketamine increases extracellular glutamate, like the mPFC.
Consistent with Autry et al. (2011), ketamine blocked NMDARs at rest in the presence of physiological concentrations of Mg2+ in mice, unlike memantine (Gideons et al., 2014). Because of that difference, ketamine reduced eEF2 phosphorylation and increased BDNF expression, while memantine did neither. Ketamine (3 mg/kg IP) had antidepressant effects in the FST and NSFT at 30 min, but memantine (3-20 mg/kg) did not. Likewise, only ketamine reduced FST immobility at 8 h and 24 h. In cultured hippocampal neurons, ketamine (50 μM), memantine, MK-801, and AP5 all reduced NMDAR-mEPSCs under magnesium-free conditions at rest, whereas in the presence of magnesium, only ketamine, AP5, and MK-801 had an effect (Gideons et al., 2014). Ketamine reduced eEF2 phosphorylation and increased BDNF in the hippocampus 30 min after injection; memantine did not. Neither drug affected eEF2 phosphorylation or BDNF at 8 or 24 h. Because the 50 μM concentration used in this study is much larger than is believed to be generated by antidepressant doses of ketamine, the results cannot be directly applied to ketamine’s antidepressant mechanism.
Miller et al. (2014) found the positive effects of ketamine on protein synthesis and depressive behavior in mice were mimicked by GluN2B deletion specifically in principal cortical neurons. GluN2B knockout (KO) mice had an increase in the ratio of currents carried by AMPARs versus NMDARs and the effect of the GluN2B-NMDAR antagonist ifenprodil on NMDAR-mediated currents was lost. Like with ketamine, loss of cortical GluN2B reduced TST and FST immobility at 30 min and 25-30 h; the reduction in both tests was large enough to occlude any additional effect of ketamine. GluN2B deletion also had an anxiolytic effect in the EPM at 30 min.
Chronic corticosterone exposure increased TST immobility in control mice, but GluN2B KO mice did not exhibit that effect, indicating less sensitivity to stress (Miller et al., 2014). Similarly, chronic stress was anxiogenic in control mice but not KO mice. In acute PFC slices prepared 24 h after ketamine (50 mg/kg IP) treatment, there was an increase in excitatory transmission onto layer II/III pyramidal neurons while at a holding potential of -65 mV and in the presence of TTX and picrotoxin, which was intended to isolate spontaneous AMPAR-mediated currents at rest. Ketamine’s effect was mimicked and occluded in GluN2B KO animals, which exhibited an increase in AMPAR-mEPSC frequency. The GluN2B-selective antagonist Ro 25-6981 also increased mEPSC events and reduced TST immobility. In contrast, chronic corticosterone treatment reduced mEPSC frequency in control mice, but not in KO mice.
Cultures prepared from GluN2B KO mouse cortex had greater protein synthesis; protein synthesis regulation depended on GluN2B, since replacement of GluN2B with GluN2A did not restore normal synthesis rates (Miller et al., 2014). Ketamine increased dendritic protein synthesis in control neurons, as did GluN2B deletion, an effect that was inhibited by rapamycin; GluN2B deletion occluded any effect from ketamine in the modified neurons. To identify the mechanism of ketamine’s effect, synaptoneurosomes were prepared from control and KO animals. In synaptoneurosomes from control mice, ketamine rapidly increased p-mTOR (a transient effect), but mTOR activity was later reduced coinciding with reduced expression of the mTOR activator Rheb, potentially implicating a negative feedback system triggered by the early increase in mTOR signaling post-ketamine. Ketamine increased synaptic protein expression, including BDNF, SAP-102, and GluA1. GluN2B KO synaptoneurosomes had increased BDNF, SAP-102, GluA1, and p-mTOR, occluding any effect of ketamine.
To confirm that GluN2B-NMDAR activation by ambient glutamate suppresses protein synthesis, control neuronal cultures were treated with TTX, DNQX, and picrotoxin to block AMPARs, GABAA receptors, and activity-dependent neurotransmitter release (Miller et al., 2014). At a holding potential of -65 mV in the absence magnesium, there was a tonic current that could be blocked by NMDAR antagonism with APV, ifenprodil, and ketamine. Cultures without the GluN2B subunit did not exhibit that current nor did cultures in which GluN2B was replaced with GluN2A. Acute cortical slices from GluN2B KO mice also did not have that current. The resting extracellular glutamate concentration was then manipulated using excitatory amino acid transporter (EAAT) inhibitors and enhancers, demonstrating that EAAT antagonism (with dl-TBOA) increased the tonic current, whereas EAAT enhancement (by NDGA) attenuated the current. Mice treated with acute NDGA or one week of ceftriaxone (another EAAT enhancer) showed an increase in mEPSC frequency in the PFC and reduction TST immobility.
The Role of AMPARs
Most ideas about how glutamatergic activity contributes to the effect of ketamine involve increased transmission through AMPARs. AMPAR activation is consistently implicated in the behavioral, biochemical, and electrophysiological effects of ketamine.
To further investigate the role of AMPARs, researchers at Yale are currently studying how the anticonvulsant perampanel, an AMPAR antagonist, interacts with ketamine in humans (NCT03367533).
Supporting Animal Research
The acute and sustained antidepressant effects of ketamine in mice are accompanied by reduced hippocampal GluR1 (Ser845) phosphorylation, which is attenuated by the AMPAR antagonist NBQX and is consistent with enhanced glutamate release (Maeng et al., 2008). In helpless LH-exposed mice, a single dose of ketamine (2.5 mg/kg IP) reduced helplessness-associated increases in escape failures and latency to escape. Impaired fear memory did not appear to mediate the effects since ketamine did not alter fear memory retention. Ketamine and imipramine reduced FST immobility at 30 min, but ketamine was distinct from imipramine in that a single dose continued to be effective two weeks later.
NBQX blocked ketamine’s effect in the FST and it also attenuated the anti-immobility effect of MK-801 and the GluN2B antagonist Ro 25,6981 in the FST at 30 min; NBQX did not alter imipramine’s anti-immobility effect (Maeng et al., 2008). Those NMDAR antagonists had less persistent effects than ketamine, with MK-801 not showing an effect one week later and Ro 25-6981 being inactive three days later. No treatment (ketamine, imipramine, NBQX) affected GluR1 protein expression, but GluR1 phosphorylation (Ser845) in the hippocampus was reduced by ketamine, a change that was blocked by NBQX; imipramine did not affect GluR1 phosphorylation.
Autry and colleagues (2011) reported that intact BDNF-TrkB signaling is necessary for ketamine’s antidepressant effects in mice. Ketamine (3 mg/kg IP) had antidepressant-like behavioral effects in control mice in the FST, NSFT, and LH tests, and it also reduced depressive behavior following CMS exposure, as seen in the SPT, NSFT, and FST. Other NMDAR antagonists (MK-801 and CPP) were also effective at 30 min and 3 h in the FST; ketamine and CPP continued to work at 24 h, but only ketamine was effective after one week.
Inducible BDNF KO mice did not respond to ketamine in the FST at 30 min or 24 h (Autry et al., 2011). Likewise, the 30 min effect of MK-801 was lost in the modified mice. TrkB KO mice were also insensitive to ketamine in the FST and NSFT, while TrkB was shown to be activated (i.e. increased receptor autophosphorylation) following NMDAR antagonist exposure in control mice. Neither ketamine nor MK-801 affected hippocampal BDNF mRNA at 30 min or 24 h, but there was an increase in BDNF protein exclusively at 30 min; ketamine also acutely (30 min) increased proBDNF. This is consistent with ketamine producing a rapid, transient increase in BDNF protein translation without altering transcription. In the cortex, but not the NAc, ketamine and MK-801 increased BDNF protein at 30 min (but not at 24 h).
Supporting the involvement of translation rather than transcription, the protein synthesis inhibitor anisomycin blocked ketamine’s effect in the FST at 30 min and 24 h, and its effect in the NSFT at 30 min (Autry et al., 2011). Anisomycin blocked mature BDNF and proBDNF synthesis in the hippocampus. Unlike anisomycin, the RNA polymerase inhibitor actinomycin D, which reduced hippocampal BDNF transcription, had no impact on ketamine’s behavioral effects at 30 min or 24 h. Ketamine increased hippocampal Arc expression, which was also attenuated by anisomycin. Hippocampal slices exposed to ketamine (20 μM) had increased AMPAR-mediated neurotransmission, although that concentration exceeds the amount generated by antidepressant doses of ketamine.
The GABAergic drug propofol did not have antidepressant effects on its own in male rats, but it enhanced the effect of ketamine (15 mg/kg IP) in the FST; the interaction seemed to involve AMPAR activity since NBQX blocked the enhancement (Wang et al., 2011). Ketamine increased hippocampal BDNF expression, but this was not enhanced by propofol. Hippocampal GluR1 phosphorylation (Ser845) was reduced in the ketamine group, whereas it was increased by propofol on its own; the propofol+ketamine group had higher Ser845 phosphorylation than the ketamine group, which would be expected to support AMPAR activity.
AMPAR antagonism with NBQX blocked the acute antidepressant-like effects of ketamine (10 or 30 mg/kg IP) in the TST (using male mice) and LH paradigm (using male rats), while the sustained effect of ketamine in the TST was partially blocked by NBQX (Koike et al., 2011). Helpless rats had more escape failures and ketamine dose-dependently attenuated that increase; it had a significant effect at 10 mg/kg, but not 3 mg/kg. In the mouse TST, immobility was reduced at 30 min exclusively at 30 mg/kg, not 3 or 10 mg/kg; acute desipramine was also effective. At 72 h, ketamine (30 mg/kg) reduced immobility, while desipramine did not, and its efficacy was attenuated by NBQX.
Ketamine had greater antidepressant-like effects in female Wistar-Kyoto (WKY) rats, which are more prone to depressive behavior, compared to Wistar rats (Tizabi et al., 2012). It dose-dependently reduced FST immobility in WKY rats, but not Wistar rats; the effect was absent at 0.5 mg/kg and appreciable at 2.5 and 5 mg/kg, causing their behavior to become roughly equivalent to the immobility level of Wistar rats. The 2.5 mg/kg dose was no longer effective one week later, but 5 mg/kg was, and by two weeks post-ketamine there was no effect from 5 mg/kg. Chronic exposure to 0.5 or 2.5 mg/kg/d for 10 days reduced FST immobility 20-22 h after the last injection and 2.5 mg/kg, but not 0.5 mg/kg, continued to reduce immobility for one week. At 0.5 mg/kg, this dosing regimen was associated with a nonsignificant reduction in NMDAR density in WKY rats (-17%) and Wistar rats (-14%), whereas there was a significant increase in AMPAR density (+26%) exclusively in WKY rats.
Lipopolysaccharide (LPS)-induced depressive behavior in male mice was attenuated by ketamine (6 mg/kg IP), with its protective effect seeming to involve NMDAR antagonism rather than inhibition of LPS-induced changes in inflammation or BDNF (Walker et al., 2013). Based on brain samples collected 24 h after LPS exposure, LPS stimulated the kynurenine pathway, producing an increase in kynurenine and an elevated kynurenine/tryptophan ratio. Downstream of kynurenine production was an increase in quinolinic acid, 3-hydroxykynurenine, and 3-hydroxyanthranilic acid, indicating the involvement of the kynurenine 3-monooxygenase pathway. LPS reduced sucrose preference and increased FST immobility; ketamine attenuated both changes. However, ketamine did not affect the LPS-induced increase in IL-6 (plasma, brain, liver), HO-1 mRNA, and plasma IL-1b. It also failed to block the increased brain and plasma kynurenine/tryptophan ratio, and the reduction in brain BDNF.
Supporting a role for NMDAR antagonism, ketamine attenuated the deficit in sucrose preference when it was administered 10 h after LPS, well after the inflammatory response had developed; ketamine also did not affect LPS-induced sickness behavior (Walker et al., 2013). NBQX blocked ketamine’s positive effect on sucrose preference.
Acute suppression of spontaneous NMDAR-mediated neurotransmission with ketamine (20 μM) potentiated synaptic responses in the CA1 region of mice and rats, and that effect was reliant on protein synthesis, BDNF expression, eEF2 activity, and elevated surface expression of GluR1/R2-containing AMPARs (Nosyreva et al., 2013). To observe the effect of NMDAR antagonism on synaptic transmission, CA3-CA1 Schaffer collateral synapses were stimulated, then stimulation ceased and ketamine (20 μM) was applied for 30 min. When stimulation resumed, ketamine potentiated field excitatory postsynaptic potentials (fEPSPs) in hippocampal slices. It selectively increased AMPAR-mediated transmission without affecting the number of synaptic inputs or NMDAR-evoked transmission.
Hippocampal slices from BDNF KO mice did not exhibit potentiation post-ketamine and in constitutive eEF2K KO mice, ketamine failed to increase hippocampal BDNF and its effects in the FST and NSFT were lost (with 5 mg/kg IP); slices from those mice did not exhibit ketamine-induced potentiation (Nosyreva et al., 2013). NASPM, a specific antagonist of GluR2-absent AMPARs, did not block ketamine-induced synaptic strengthening. In contrast, the subunit-independent AMPAR blocker DNQX attenuated ketamine-induced potentiation. Consistent with those results, hippocampal slices from GluR2 KO mice did not exhibit a change in synaptic strength post-ketamine and GluR2 KO mice did not have acute antidepressant effects in the FST and NSFT.
Acute FST immobility reduction 30 min after ketamine (10 mg/kg IP) is accompanied by increased BDNF and p-mTOR in the PFC and hippocampus of male rats (Zhou et al., 2014). NBQX attenuated the reduction in FST immobility and the AMPAR positive modulator CX546, which was ineffective on its own, enhanced ketamine-induced immobility reduction. AMPAR antagonism with NBQX also attenuated the increase in p-mTOR and BDNF, whereas CX546 significantly enhanced ketamine’s induction of p-mTOR and BDNF in the PFC, as well as BDNF in the hippocampus, with a nonsignificant enhancement of hippocampal p-mTOR.
Chronic (11-day) AMPA itself reduces FST immobility in male rats, increases sucrose preference, and elevates hippocampal BDNF, synapsin I, and p-mTOR (Akinfiresoye and Tizabi, 2013). Those effects were also present when chronic subtherapeutic ketamine (0.25 mg/kg IP) was combined with subtherapeutic AMPA, yielding increased sucrose preference and reduced FST immobility 20-22 h after the final ketamine dose and 20 min after the last AMPA injection. Both drugs were individually effective at 0.5 mg/kg and became somewhat more effective when combined, although the combo was not substantially superior in most cases. The effects were relatively short-lasting, with no behavior change one week after treatment.
FST immobility was reduced by ketamine (10 mg/kg IP) and the mGluR2/3 antagonist LY341495 in male rats at 24 h; NBQX attenuated the effect of both drugs (Koike and Chaki, 2014).
Fukumoto et al. (2016) reported that the antidepressant effects of ketamine and LY341495 are associated with activation of dorsal raphe nucleus (DRN) serotonin neurons, which seems to be dependent on AMPAR stimulation in the mPFC of male mice. At 30 min, FST immobility was reduced by LY341495, ketamine (30 mg/kg IP), and the SSRI paroxetine; serotonin depletion with PCPA blocked the effects. LY341495 and ketamine also reduced immobility at 24 h, but paroxetine was ineffective, and those changes were similarly blocked by serotonin depletion. Immobility at 30 min and 24 h was also reduced by intra-mPFC injection of ketamine or LY341495; serotonin depletion attenuated that effect. Systemic or intra-mPFC injection of NBQX blocked the FST immobility reduction caused by both drugs. Intra-mPFC ketamine and LY341495 increased the proportion of DRN serotonin cell bodies exhibiting c-Fos immunoreactivity (an indicator of activity), which was blocked by intra-mPFC NBQX.
Ketamine potentiated fEPSPs induced by stimulation of Schaffer collateral pathways in rat hippocampal slices with an EC50 of 53 nM, as measured in the CA1 stratum radiatum region; pretreatment with the protein kinase A (PKA) inhibitor H89 blocked ketamine’s effect (Zhang et al., 2016). Ketamine increased GluR1 phosphorylation (Ser845) in the CA1 and increased total GluR 15 min after exposure; H89 blocked the increase in GluR1 phosphorylation, but not the change in GluR1 abundance. The protein synthesis inhibitor anisomycin blocked the ketamine-induced increase in GluR1 without affecting SC-CA1 fEPSPs or GluR1 phosphorylation. TrkB inhibition with K252a attenuated the increase in GluR1, whereas H89 and the CaMKII inhibitor KN62 were ineffective. At SC-CA1 synapses, ketamine increased GluR1 surface abundance, which was blocked by H89 despite H89 not affecting total GluR1 abundance.
In male rats, FST immobility was reduced by ketamine (10 mg/kg IP) and GluR1 abundance and phosphorylation increased within 30 min (Zhang et al., 2016). When hippocampal slices were treated with picrotoxin (GABAA inhibitor) and CGP52432 (GABAB inhibitor) to block inhibitory transmission, ketamine still enhanced SC-CA1 EPSCs in a PKA-dependent manner; the same occurred with MK-801. GABA receptor inhibition also did not block ketamine’s induction of GluR1 abundance and phosphorylation.
A serine-to-alanine mutation of the GluR1 gene, which prevents phosphorylation by PKA, prevented ketamine-induced enhancement of SC-CA1 fEPSPs in mouse hippocampal slices and it did not increase surface abundance of GluR1 (Zhang et al., 2016). That mutation also prevented FST immobility reduction in mice. Antagonism of presynaptic CA3 neurons seems to be important for ketamine’s effect rather than inhibition of NMDARs on postsynaptic CA1 neurons. SC-CA1 fEPSPs were enhanced by ketamine and antidepressant effects were present in mice with NMDARs knocked out on CA1 neurons, but ketamine was ineffective in mice with knockout of NMDARs on CA3 neurons.
Relevant Ketamine Research
Low doses of ketamine (10-30 mg/kg IP) increase extracellular glutamate and dopamine in the PFC of rats, whereas an anesthetic dose (200 mg/kg) reduces glutamate and an intermediate dose (50 mg/kg) has no effect (Moghaddam et al., 1997). Intra-PFC application of the AMPAR antagonist CNQX attenuated the PFC dopamine increase, as did systemic treatment with the AMPAR antagonist LY293558. There was a trend towards elevated glutamate in the striatum, but the impact was highly variable, although there was an increase in dopamine. At 30 mg/kg, ketamine impaired spatial delayed attention performance, an effect attenuated by AMPAR antagonism with LY293558.
Rats treated with ketamine (10 mg/kg IP) exhibit an increase in AMPAR-mediated currents in pyramidal neurons one day later (Bjorkholm et al., 2015). In vitro research using layer V/VI pyramidal neurons of the mPFC found a combination of fluoxetine and olanzapine also increased AMPA-induced currents at 30 min, but not 5 min; they were ineffective on their own. The effect of that combo was prevented by the D1 receptor antagonist SCH23390. Olanzapine also caused a small increase in NMDA-induced current at 5 min, but not 30 min, and there was a small increase from fluoxetine at 30 min; in contrast, the combo enhanced NMDA-currents at both timepoints and their impact could be blocked by SCH23390.
In an AMPAR-dependent manner, ketamine increased catecholaminergic activity in male rats receiving 10 or 25 mg/kg (IP) (El Iskandrani et al., 2015). Acute exposure increased dopamine neuron population activity (i.e. the number of active neurons per electrode) by 113%, which was blocked by NBQX, but it did not affect the firing rate of dopamine neurons; ketamine (10 or 25 mg/kg) did not affect DRN serotonin neuron firing. Ketamine also acutely increased the average rate of locus coeruleus noradrenergic neuron firing and nearly doubled the proportion of noradrenergic neurons exhibiting burst activity, effects that were blocked by NBQX. Ketamine (10 or 25 mg/kg) did not affect NMDA-evoked firing activity in hippocampal CA3 pyramidal neurons, but at 30 min there was a 64% increase in AMPA-induced firing activity.
Relevant Non-Ketamine Research
In Vitro
The AMPAR positive modulators CX614 and CX546 increased BDNF mRNA and protein expression in cultured rat entorhinal/hippocampal slices; those changes were associated with an increase in synchronous discharges (Lauterborn et al., 2000). AMPAR antagonists suppressed the increase, but NMDAR antagonists did not. L-type VDCC blockade also prevented induction in the entorhinal cortex, but not in the hippocampus. With a prolonged infusion of the positive modulators, BDNF mRNA levels peaked at 12 h and returned to baseline within two days, whereas BDNF protein remained elevated at least through the 48-hour incubation period. NGF mRNA was increased, but it returned to baseline more quickly. Consistent with these results, in vivo research with rodents showed an increase in hippocampal BDNF mRNA following CX546 exposure.
AMPAR potentiation with LY392098 reduced FST immobility in rats and mice, and it also reduced TST immobility (Li et al., 2001). The noncompetitive AMPAR antagonist LY300168 blocked its efficacy in the FST. In contrast, AMPAR antagonism did not attenuate the antidepressant-like effects of imipramine.
Antidepressant-like effects were reported with the selective group II mGluR antagonist MGS0039 in the TST at 20 min using male mice and that effect was blocked by NBQX (Karasawa et al., 2005). Treatment also increased mPFC serotonin levels, which was attenuated NBQX.
Two drugs with potential efficacy in depression, lamotrigine and riluzole, increased GluR1 and GluR2 expression after three days of treatment in cultured hippocampal neurons; in contrast, valproate reduced GluR1 (Du et al., 2007). When hippocampal neurons exposed to lamotrigine or riluzole for three days were stimulated by AMPA, AMPA-induced depolarization was potentiated. Phosphorylation of GluR1 at the PKA site (Ser845) is important for receptor insertion into the neuron surface; phosphorylation was increased by lamotrigine and riluzole, whereas valproate reduced phosphorylation. In mice treated with lamotrigine, riluzole, or imipramine for 10 days, hippocampal GluR1 (Ser845) phosphorylation increased.
Exposure to CMS produced anhedonia in adult rats, as assessed by the SPT, but it did not have that effect in young male rats (Toth et al., 2008). The anhedonic effect of stress was associated with reduced hippocampal neurogenesis, reduced hippocampal BDNF, reduced hippocampal GluR1 expression, and increased GluR1 in the anterior NAc of adult, but not young rats. There was a reduction in GluR1 expression in the PFC of both young and adult rats, no change in the posterior NAc, reduced GluR1 in the dorsal DG of adults, and no significant effects in the ventral DG, anterior VTA, or posterior VTA.
General Role of Glutamatergic Activity
Humans
Ketamine increased glutamate in the ACC of 13 healthy males at a plasma concentration of 150 ng/mL, as assessed by 1H-MRS 25 min after the start of the infusion (Stone et al., 2012). It increased PANSS ratings for positive, negative, and general schizophrenic-like effects; glutamate ACC correlated with positive symptom scores, but not the negative or general subscales. This concentration of ketamine did not affect subcortical GABA levels.
DeLorenzo et al. (2015) tested ketamine’s effect on the binding of an mGluR5 ligand, ABP688, which would be expected to drop in the presence of increased extracellular glutamate. Indeed, in 10 healthy people who received ketamine (0.23 mg/kg bolus, then 0.58 mg/kg over 1 h), binding was reduced by 20-25% in the ACC, mPFC, OFC, parietal lobe, dorsal putamen, dorsal caudate, amygdala, and hippocampus. Binding in the cerebellum was reduced by an average of 16%, but the effect ranged from a 42% reduction to a 14% increase. The participants experienced dissociative effects (CADSS), but there was no correlation between CADSS subscales (average, amnesia, depersonalization, or derealization) and ketamine blood level or ABP688 binding.
Animals
Glutamate and dopamine release were increased in the mPFC of male rats after ketamine (18 mg/kg SC) administration (Lorrain et al., 2003). Those effects were blocked by systemic and intra-mPFC administration of the mGluR2/3 agonist LY379268 and by intra-mPFC TTX. Intra-mPFC ketamine did not affect glutamate release, but it increased dopamine.
Chowdhury et al. (2012) reported an increase in 13C-enrichment of glutamate, GABA, and glutamine in the mPFC of male rats given ketamine (30 mg/kg IP) and evaluated using MRS. A quantitative increase in neurotransmitter cycling was detected in the hippocampus, but it was nonsignificant. Ketamine’s effect in the mPFC was lost at an anesthetic dose (80 mg/kg).
At intermediate doses (3 and 10 mg/kg), glutamate, GABA, and glutamine cycling were increased by ketamine, as determined by infusing 13C-labeled glucose and measuring 13C enrichment in dissected mPFC (Chowdhury et al., 2016). Glutamate enrichment was elevated at the 10-18 min timepoint, preceding its antidepressant effects. Ketamine reduced FST immobility at 24 h when given at 30 mg/kg, but not 3 or 80 mg/kg. Ketamine-like effects were also seen with the GluN2B-NMDAR antagonist Ro 25,6981. Ketamine did not affect glutamate cycling at 1 mg/kg (IP), but at 30 mg/kg it increased glutamate enrichment (+9%) and glutamine enrichment (+38%), whereas there was no effect at 80 mg/kg. That dose-response profile is consistent with its antidepressant effects, which are lost at higher doses.
Ketamine and norketamine, both of which primarily exhibit NMDAR antagonism, had antidepressants effects in the mouse FST at 30 min, with a minimum effective dosage of 10 mg/kg (IP) for ketamine and 50 mg/kg for norketamine (Salat et al., 2015). In contrast, the weaker NMDAR antagonist DHNK was ineffective in the FST with up to 50 mg/kg. Neither ketamine nor norketamine (50 mg/kg of each) reduced immobility on Days 3 or 7; rather, ketamine-treated mice had an increase in immobility one week later.
Research Suggestive of Non-NMDAR Activity
Ketamine and its metabolite (2R,6R)-HNK increased intracellular cAMP, p-CREB, and BDNF expression via an NMDAR-independent mechanism (Wray et al., 2019). C6 cells treated with ketamine (10 μM) for 15 min or 24 h showed an increase in Gαs liberation from lipid rafts within 15 min; because 15 min was adequate for inducing this effect, that exposure time was used for subsequent experiments. The translocation of Gαs caused by 15 min of ketamine exposure was similar to the effect of repeated standard antidepressant exposure. Because 10 μM is a fairly large concentration, the effects of 1-10 μM were also tested. There was a dose-dependent increase in Gαs movement from lipid rafts, with a significant effect observed by 1 μM. With 10 μM, translocation was detected for at least 12 h, but it was back to baseline 24 h after exposure.
Other NMDAR antagonists (MK-801, memantine, and AP-V) failed to replicate ketamine’s effect, suggesting a different target is involved. MOR agonism and KOR antagonism were ruled out, as DAMGO and nor-BNI did not affect Gαs localization (Wray et al., 2019). cAMP was measured in live cells; basal fluorescence was the same in control and ketamine-treated cells, but when ketamine-treated cells were stimulated by the Gαs-coupled agonist isoproterenol, they exhibited larger and faster increases in fluorescence, indicative of increased coupling of Gαs and adenylyl cyclase.
When the obligatory GluN1 NMDAR subunit was knocked down, ketamine had a similar effect on cAMP, further supporting an NMDAR-independence mechanism (Wray et al., 2019). Because cAMP activates PKA, resulting in phosphorylation of CREB, and p-CREB functions as a transcription factor in the nucleus for genes relevant to cellular survival, growth, and synaptic plasticity, the impact on CREB was assessed. Ketamine induced a sustained increase in p-CREB at 15 min and 24 h, like other antidepressants, which have been shown to increase p-CREB after chronic exposure.
C6 cells treated with ketamine (10 μM) for 15 min had a sustained increase in p-eEF2 (at Thr56), while total eEF2 level was unaffected (Wray et al., 2019). Ketamine increased BDNF via a cAMP/PKA/CREB-mediated transcription-dependent route at 24 h, but not at 1 h. When cAMP was antagonized using Rp-cAMPS in rat astrocytes treated with ketamine, the ketamine-induced increase in BDNF at 24 h was attenuated. This suggests ketamine increases BDNF production from astrocytes in a cAMP-dependent manner. Interestingly, (2R,6R)-HNK, which is not an NMDAR antagonist at typical concentrations, had ketamine-like effects following 15 min of exposure at 10 μM; Gαs lipid raft localization was reduced and cAMP was increased (Wray et al., 2019).
Relevant Non-Ketamine Research
Humans
Using postmortem samples from depressed (n=9) and healthy people (n=9), Karolewicz et al. (2005) reported higher GluN1 and GluN2C NMDAR subunit expression in the locus coeruleus of depressed subjects; there was only weak immunoreactivity for GluN2A and GluN2B in this region. GluN1 expression in the cerebellum did not differ between the groups, but GluN2C expression was nonsignificantly increased in depressed subjects (+35%).
The total amount of glutamate/glutamine (Glx) in the dorsomedial/dorsal anterolateral PFC and in the ventromedial PFC (vmPFC) was reduced in depression, as determined with 1H-MRS scans of unmedicated MDD patients (n=20) compared to controls (n=20) (Hasler et al., 2007). GABA was also reduced in the dorsomedial/dorsal anterolateral PFC. MRS does not distinguish between extracellular versus intracellular neurotransmitters and there is a 50- to 100-fold greater intracellular glutamate pool compared to the extracellular concentration (Featherstone and Shippy, 2007), so differences observed with MRS cannot be simply interpreted as altered glutamate release. Instead, these results could be reflective of the glial loss that occurs in depression, which may impair glutamate accumulation over time.
Psychiatric conditions including schizophrenia, BD, and MDD have been associated with altered glutamate receptor function and expression (Beneyto et al., 2007). Based on postmortem brain samples from subjects with schizophrenia (n=15), MDD (n=15), or BD (n=15), as well as from control subjects (n=15), all psychiatric conditions were associated with reduced perirhinal cortex GluR5 expression. Additionally, GluR1, GluR3, and GluN2B were reduced in the perirhinal cortex of BD and MDD subjects; GluN2A was reduced in MDD. In the entorhinal cortex, BD was associated with reduced GluR2, GluR3, and GluR6 mRNA expression.
Binding of the NMDAR antagonist MK-801 was reduced in the hippocampus of schizophrenia and BD subjects, while binding of CGP39653 (glutamate site on NMDAR) was increased in MDD subjects and binding of MDL105,519 (glycine site of NMDAR) was increased in BD subjects (Beneyto et al., 2007). Hippocampal AMPAR and kainate receptor changes were not observed.
NMDAR subunit expression was reduced in postmortem anterior PFC samples from MDD patients (n=14) compared to controls (n=10) (Feyissa et al., 2009). MDD subjects did not have psychotropic medications in their system at the time of death. The reduction in subunit expression was specific to GluN2A (-54%) and GluN2B (-48%), with no difference in GluN1. The postsynaptic protein PSD95 was also reduced by 40%.
Duric et al. (2013) reported dysregulation of AMPAR subunits in postmortem brains from MDD (n=21) subjects compared to controls (n=18). Downregulation of the glutamate receptor genes GLUR1 and GLUR3 was present in the DG and CA1 of patients, and GLUR4 was reduced in the DG. Some presynaptic genes were affected in the hippocampus, with a reduction in synapsin 3 and synaptosomal-associated protein 25 kDa (SNAP25); more postsynaptic genes were affected, with downregulation of synapse-associated proteins (SAP) 93 and 102, along with microtubule-associated proteins 1a, 1b, 2, and t (encoded by MAP1A, MAP1B, MAP2, and MAPT). Serotonin receptor genes were altered in the hippocampus, specifically with overexpression of HTR2C in the DG but not CA1, and HTR4 and HTR7 were downregulated in the DG and CA1.
Animals
In male mice, chronic (14-day) exposure to the SSRI citalopram reduces the proportion of high-affinity, glycine-displaceable binding sites for the NMDAR antagonist CGP-39653 and reduces the potency of glycine for inhibiting the binding of DCKA (a glycine-site NMDAR antagonist) in the cortex but not the hippocampus (Nowak et al., 1996). Citalopram also increased aspartate levels in the cortex and hippocampus.
Prolonged stress has a degenerative effect (e.g. dendritic atrophy) in the hippocampus that is blocked by NMDAR antagonists (McEwen, 1999). Potentially mediating the effect of chronic stress, adrenal glucocorticoid hormones alter NMDAR expression in the hippocampus and increase NMDAR binding; glucocorticoids also suppress neurogenesis in an NMDAR-dependent manner. The negative effects of stress, which is known to acutely increase glutamate release, are attenuated by inhibitors of glutamate release. Interestingly, the serotonin system influences glutamatergic activity, with serotonin potentiating NMDAR binding and activity, an effect that may involve 5-HT2 receptors.
Chronic restraint or unpredictable stress in male rats impaired PFC-mediated temporal order recognition memory, which is accompanied by reduced AMPAR- and NMDAR-mediated synaptic transmission, glutamate receptor expression on PFC pyramidal neurons, and mEPSC amplitude (Yuen et al., 2012). The loss of glutamate receptors involved glucocorticoid receptor activation, which produced ubiquitin-proteasome degradation of GluR1 and GluN1 subunits. Proteasome inhibition prevented memory impairment and the reduction in glutamatergic activity. Further supporting the role of glucocorticoids, the effect of chronic stress was replicated by repeated injections of corticosterone into the PFC and the impact of restraint stress was blocked by the glucocorticoid receptor antagonist RU486. Unlike chronic glucocorticoid exposure, acute corticosterone increased mEPSC amplitude.
7.2 mTOR
Mammalian target of rapamycin (mTOR) is a key regulator of many physiological processes in the body. As a protein kinase, it alters protein structure through the addition of phosphoryl groups to regulate activity; mTOR signaling is known to affect protein synthesis and is involved in synaptic plasticity in the brain.
There are two mTOR complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 is the most well-studied and details about its activity and structure were discovered first. Using human embryonic kidney (HEK) cells, Kim and colleagues (2002) found mTOR forms a complex with a protein called raptor (regulatory associated protein of mTOR) as well as GβL (aka MLST8). The mTOR-raptor complex plays a key role in nutrient-sensing; when activated, it affects protein synthesis and cellular maintenance. Those effects partly come from the downstream effector p70S6K (p70 ribosomal protein S6 kinase), which phosphorylates the ribosomal protein S6 to induce protein translation.
Much of what we know about mTOR comes from early studies investigating the effects of rapamycin, an immunosuppressant and anticancer drug. That research led to the discovery of mTOR, and mTOR’s function has been studied for decades using rapamycin because it destabilizes the mTOR-raptor association, making it an ‘mTOR inhibitor.’ That complex is now known as mTORC1; as such, rapamycin is an mTORC1 inhibitor. Its inhibitory effect is indirect since rapamycin’s actual receptor is FKBP12, but the resulting FKBP12-rapamycin complex binds to mTOR and interrupts its activity.
The mTOR-raptor complex senses nutrients and alters intracellular activity to control cell growth and maintenance. mTORC1 activity, cell growth, and protein synthesis are upregulated when certain nutrients (e.g. amino acids) are present because those nutrients reflect a well-fed nutritional state in the organism.
mTOR does not exclusively interact with raptor. Sarbassov et al. (2004) studied mammalian cells and discovered mTOR also forms a distinct complex with rictor (rapamycin-insensitive companion of mTOR) and GβL. Rictor was confirmed to be a homolog of a protein from S. cerevisiae (yeast) called AVO3p, which formed a TOR complex that was not affected by rapamycin. Instead of signaling through p70S6K like mTORC1, this complex—known as mTORC2—regulates protein kinase C alpha (PKCα) activity to affect the actin cytoskeleton.
The effects of ketamine seem to involve mTORC1 considering the importance of mTORC1 signaling in protein synthesis regulation—upregulation of synaptic protein synthesis is consistently seen with ketamine—and rapamycin’s ability to inhibit many of the antidepressant-relevant effects of ketamine in animals and in vitro (Li et al., 2010, 2011).
Supporting Research
Humans
A female TRD patient treated with S-ketamine (0.25 mg/kg IV, 40-min) had an increase in mTOR phosphorylation in peripheral blood cells (Denk et al., 2011). mTOR phosphorylation was increased up to 100 min after the start of the infusion.
In three male MDD patients treated with ketamine (0.5 mg/kg), plasma p-mTOR increased and there was a trend towards increased p-eEF2 (Yang et al., 2013). The increase was observed at 10 min through 120 min. In addition to the elevation in mTOR and eEF2 phosphorylation, p-GSK-3β increased.
Animals
In 2010, Nanxin Li and colleagues at Yale University demonstrated rapamycin inhibition of ketamine’s antidepressant-like effects in male rats when it was administered to the brain through the intracerebroventricular (ICV) route or when administered directly to the mPFC (Li et al., 2010). Ketamine (10 mg/kg IP) rapidly activated the mTORC1 pathway in the PFC, producing increased phosphorylation of the mTORC1 effectors 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein 1) and p70S6K. Phosphorylation of mTOR was increased, consistent with its activation. This effect of ketamine was unique among antidepressants, as ECT, imipramine, and fluoxetine all failed to acutely increase mTORC1 signaling. Li et al. also showed the effect was highly dose-dependent, occurring at 5-10 mg/kg, but disappearing at larger anesthetic doses. Along with increased activation of mTORC1 effectors, phosphorylation of extracellular signal-related kinases (ERKs) and protein kinase B (PKB; Akt) was increased in the PFC.
Akt sits upstream of mTORC1, so the effect of ERK and Akt inhibition was tested (Li et al., 2010). When ERK was inhibited by U0126 or phosphoinositide 3-kinase (PI3K)—Akt is downstream of PI3K—was inhibited by LY294002, the ketamine-induced increase in mTOR, 4E-BP1, and p70S6K phosphorylation was blocked. Given the role of mTORC1 signaling in protein synthesis, the effect of ketamine on synaptic proteins was studied using synaptoneurosomes from rat PFC. Ketamine (10 mg/kg IP) increased levels of the presynaptic protein synapsin I and the postsynaptic proteins PSD-95 (postsynaptic density protein 95) and GluR1, a subunit of AMPARs; those changes were delayed compared to mTORC1 activation. While phosphorylation of mTORC1 and its effectors occurred shortly after administration and returned to baseline within a couple hours, synaptic protein levels peaked at 2-6 hours and remained above baseline for up to three days, perhaps functioning as one of the mechanisms that enables a short-acting drug like ketamine to have lasting effects for 3-7 days. Rapamycin blocked ketamine’s induction of synaptic proteins.
The mTORC1-dependent increase in synaptic proteins was accompanied by an increase in dendritic spine formation and synaptogenesis in mPFC pyramidal neurons, potentially increasing the capacity for connections between neurons (Li et al., 2010). Importantly, that increase occurred 24 h after ketamine exposure, meaning it was present after ketamine had already been cleared from the body. Synaptic strength and spine maturation were elevated by ketamine, as shown by an increase in ‘mushroom’ spines (i.e. spines with a larger head). Increased synaptic strength correlated with an increase in serotonin- and orexin-induced EPSC frequency.
A role for mTORC1 in the antidepressant mechanism was further shown using behavioral mood-relevant tests, namely the FST, LH, and NSFT (Li et al., 2010). Some traditional antidepressants are acutely active in the FST, but for the other tests, repeated (6-21 days) exposure is required, similar to the efficacy delay in humans. Differentiating itself from those drugs, ketamine was acutely effective in every test. Rapamycin (ICV or intra-mPFC) blocked its effect in the FST and NSFT. As with the cellular and molecular effects of ketamine, its behavioral effects were attenuated by inhibition of ERK (with U0126) or PI3K/Akt (with LY294002).
Ketamine (10 mg/kg IP) increased synaptic protein levels in the PFC of rats, thereby supporting synaptic plasticity, according to Li et al. (2011). Chronic stress via the CUS paradigm produced the opposite effect, causing a deficit in synaptic protein levels (GluR1, PSD-95, and synapsin I), spine number, and the frequency/amplitude of serotonin- and orexin-induced EPSCs in layer V pyramidal neurons of the PFC. Those deficits were reversed by one dose of ketamine when tested 24 h after drug exposure; rapamycin inhibited ketamine’s beneficial effect. When tested one week after administration, ketamine stilled attenuated the synaptic protein deficit caused by stress. Behaviorally, ketamine was effective one week later in the SPT despite continued CUS exposure. Ketamine’s effects were largely replicated by the GluN2B-NMDAR antagonist Ro 25,6891, including in the SPT one week after drug administration.
Koike et al. (2011) found the antidepressant-like effect of ketamine (30 mg/kg IP) in male mice subjected to the TST 24 h after exposure was attenuated by rapamycin administered via the ICV route. Similarly, rapamycin blocked the antidepressant-like effect of the mGluR2/3 antagonists LY341495 and MGS0039 in the TST at 24 h. In contrast, rapamycin did not block the antidepressant effect of LY341495 at 30 min. NBQX attenuated the 30 min effect of LY341495 and partially blocked its 24 h effect.
Male mice exposed to a CMS paradigm had a reduction in sucrose preference in the SPT and increased immobility in the TST and FST (Tang et al., 2015). Additionally, CMS reduced phosphorylation of the mTORC1 effectors 4E-BP1 and p70S6K in the PFC, and it reduced phosphorylation of Akt and ERK1/2 in the PFC. Ketamine (30 mg/kg IP) had antidepressant-like effects in CMS-exposed mice for up to five days, reversing sucrose preference reduction within two hours, an effect that was maintained for two days, though its efficacy was lost by Day 6. Ketamine reduced immobility in the TST and FST for five days and reversed a deficit in food consumption in the NSFT, though only until Day 2, not Day 6.
Two days after treatment, ketamine reversed the CMS-induced reduction in 4E-BP1 and p70S6K phosphorylation, along with reversing the Akt and ERK1/2 phosphorylation deficit (Tang et al., 2015). In evaluating the effects of stress on the glutamatergic system, the authors observed an increase in GluN1 expression in the PFC, which was reversed on Day 2 by ketamine. Neither ketamine nor stress affected the expression of the GluN2A and GluN2B subunits. CMS reduced expression of the AMPAR GluR1 subunit as well as expression of the synaptic proteins PSD95 and synapsin I; those deficits were reversed by ketamine on Day 2. This demonstrates that ketamine causes sustained alleviation of chronic stress-induced changes in prefrontal ERK/Akt signaling, mTORC1 signaling, and synaptic protein expression; it is effective for a number of days, although most of its effects are lost by Day 6.
Research by Harraz and colleagues (2016) demonstrated that the mechanism underlying ketamine’s mTOR activation may involve the inhibition of NO formation, which reduces nitrosylation of GAPDH, thereby inhibiting the formation of the GAPDH/Rheb/Siah1 complex that would normally reduce Rheb and, as a result, mTOR activity. In primary cortical cultures from mice, NMDA rapidly reduced Rheb as well as the formation of p-p70S6K, whereas ketamine increased Rheb and p-p70S6K. The GluN2B inhibitor CERC-301 (MK0657) mimicked ketamine, but memantine did not affect Rheb expression or mTORC1 pathway activity. Similar results were reported in vivo; mice given ketamine (10 mg/kg IP) had a rapid increase in Rheb and p-p70S6K in the hippocampus.
The NMDA-induced reduction in Rheb in vitro appeared to involve proteasome-mediated degradation since the proteasome inhibitor clasto-lactacystin β-lactone blocked the NMDA-induced Rheb and p-p70S6K reduction (Harraz et al., 2016). NMDA and the NO donor CysNo increased GAPDH-Rheb binding, which was inhibited by CGP3466B, an inhibitor of GAPDH nitrosylation. Involvement of NO was further supported by the observation that the nitric oxide synthase (NOS) inhibitor nitroarginine and the nNOS-specific inhibitor 7-nitroindazole also blocked NMDA’s effect, indicating NMDAR signaling increases GAPDH-Rheb binding through a mechanism involving NO, which can increase nitrosylation of GAPDH and promote its association with Rheb. NMDA’s stimulation of GAPDH-Rheb binding correlated with a reduction in p-p70S6K. In contrast, ketamine reduced GAPDH-Rheb binding. NMDA also stimulated Siah1-Rheb binding, which involved GAPDH in the presence of NO.
Overall, the research by Harraz et al. (2016) suggests NMDAR activation increases NO formation; from there, NO nitrosylates GAPDH, enhancing its binding to Siah1 and Rheb. When Siah1 is depleted via shRNA, Rheb is increased in vitro, demonstrating the ubiquitin-E3-ligase activity of Siah1, which causes the degradation of Rheb. When Rheb is depleted, mTOR signaling is reduced. By inhibiting NMDAR, ketamine causes the opposite of these effects, ultimately elevating mTORC1 activation. Consistent with this mechanism, CGP3466B, an inhibitor of GAPDH nitrosylation, had ketamine-like effects in the FST and NSFT; like with ketamine, rapamycin blocked the behavioral effects of CGP3466B.
An effect on NO was also identified by Zhang et al. (2013) who reported that the FST immobility reduction caused by ketamine (10 mg/kg IP) in male rats at 60 min correlated with a reduction in hippocampal total NOS (T-NOS), iNOS, and eNOS. Plasma activity of T-NOS, iNOS, and eNOS was increased in the ketamine group, while nNOS was not affected by ketamine in plasma or the hippocampus. The antidepressant effect of ketamine was blocked by pretreatment with L-arginine, an NO precursor. Conversely, subtherapeutic ketamine (3 mg/kg IP) became effective when combined with a subtherapeutic dose of the NOS inhibitor L-NAME. This suggests NO inhibition may contribute to ketamine’s antidepressant effects.
Because most studies have used racemic ketamine, differences in mTORC1 activity between the enantiomers have largely gone unexamined. A paper from Yang and colleagues addressed this in 2018. They found the antidepressant effect of S-ketamine (10 mg/kg IP) in CSDS-susceptible mice seems to involve mTOR activation, whereas R-ketamine (10 mg/kg IP) is reliant on the ERK pathway (Yang et al., 2018). Rapamycin (ICV) blocked the behavioral effects of S-ketamine in the TST and FST one day after drug exposure, as well as SPT behavior after one week, but it failed to alter the effects of R-ketamine. Similarly, the mTOR inhibitor AZD8055 only blocked the antidepressant-like effects of S-ketamine.
CSDS-susceptible mice had a reduction in p-mTOR in the PFC, CA3, and DG, but not in the CA1; there was an increase in the NAc (Yang et al., 2018). S-ketamine attenuated the reduction in p-mTOR in the PFC, CA3, and DG, yet R-ketamine enhanced the reduction in the DG; neither enantiomer affected mTOR activation in the NAc. Consistent with these findings, susceptible mice had a reduction in p-p70S6K in the PFC, though not in the NAc, CA1, CA3, or DG. Only S-ketamine attenuated this reduction. The behavioral effects of R-ketamine in the TST, FST, and SPT were blocked by the ERK inhibitor SL327, which had no effect on S-ketamine.
GluR1 and BDNF were reduced in the PFC of susceptible mice, but neither ketamine enantiomer reversed this after 30 min (Yang et al., 2018). CSDS reduced p-ERK in the PFC, CA3, and DG, but not in the CA1, and it increased p-ERK in the NAc; this was only attenuated by R-ketamine (tested at 30 min), although its effects were limited to the PFC, CA3, and DG, with no impact in the NAc. CSDS also reduced p-MEK in the PFC and DG, but not in the NAc, CA1, or CA3; only R-ketamine attenuated the reduction in the PFC and DG of susceptible mice.
Opposing Research
Humans
In a DBRCT crossover trial of 20 depression patients, rapamycin (6 mg oral) administered two hours before ketamine (0.5 mg/kg IV, 40-min) did not impair its antidepressant effects (Abdallah et al., 2020). The effect of ketamine at 24 h was unaffected and, surprisingly, its efficacy in the two-week period after administration was enhanced by rapamycin. Two weeks after ketamine, the response rate was higher in those who received rapamycin (41% vs. 13%); likewise, the remission rate was increased in the rapamycin group (29% vs. 3%).
Animals
Autry et al. (2011) reported that rapamycin did not block the antidepressant-like effects of ketamine (3 mg/kg IP) in the FST, NSFT, and LH tests at 30 min in mice. This was despite phosphorylation of p70S6K being reduced in the cortex and hippocampus, which confirmed systemic rapamycin was active in the brain. The rapid effect of ketamine was independent of altered mTOR phosphorylation in control and BDNF KO hippocampal tissue and in WT cortical tissue.
Since many studies have shown an effect of rapamycin at 24 h or days after ketamine exposure, this study does not necessarily contradict that research. Instead, mTOR signaling could be important for the persistent antidepressant effects of ketamine, whereas other mechanisms are responsible for its acute efficacy.
Relevant Research
In male rats, ketamine (40 mg/kg IP), norketamine (20 mg/kg), and (2S,6S)-HNK (20 mg/kg) increased phosphorylation of mTOR and its downstream effectors (Paul et al., 2014). The increase in p-mTOR and p-p70S6K peaked at 30-60 min. There were only nonsignificant increases in p-4E-BP1, p-ERK1/2, and p-Akt. The mTOR pathway was also activated in vitro, as shown in PC-12 cells, where the expression of monomeric serine racemase (m-SR) was increased 2-fold by (2S,6S)-HNK (0.05 nM), norketamine (10 nM), and ketamine (100 nM).
The peak m-SR increase in PC-12 cells occurred with 600 nM ketamine, 10 nM of norketamine, and 0.05 nM of (2S,6S)-HNK (Paul et al., 2014). Each drug increased phosphorylation of mTOR, 4E-BP1, p70S6K, ERK1/2, and Akt in vitro, specifically with 400-600 nM ketamine, 1-250 nM norketamine, and 0.01-10 nM of (2S,6S)-HNK. Those effects were greatly reduced or eliminated at higher concentrations, particularly >2000 nM of ketamine, >250 nM of norketamine, and >1 nM of (2S,6S)-HNK.
7.3 Monoamines
The monoamine theory of depression has been highly influential in medicine and in the general public’s understanding of depressive disorders. Although research has consistently shown antidepressants have more influential mechanisms than simple monoamine modulation, that does not exclude a role for serotonin, norepinephrine, and dopamine in the pathophysiology of depression or the mechanism of antidepressants, including ketamine.
Serotonin
Multiple studies have demonstrated an apparent role for serotonin in ketamine’s antidepressant effects (Gigliucci et al., 2013; Nishitani et al., 2014; Fukumoto et al., 2014; Pham et al., 2017). Instead of ketamine relying on direct serotonin receptor or transporter activity, it may depend on intact neurocircuitry that uses serotonin
In male rats, three days of serotonin depletion using the tryptophan hydroxylase inhibitor PCPA eliminated ketamine’s antidepressant-like effect in the FST 24 hours after administration, but it did not prevent its acute effect at 1 h (Gigliucci et al., 2013). To induce depressive behavior, rats were exposed to 10 days of repeated immobilization stress. FST immobility was increased by chronic stress, while ketamine reduced immobility at 1 h with 25 mg/kg (IP), but not with 10 mg/kg. There was no observable effect on frontal cortex serotonin levels from ketamine itself, but the ~75% reduction in frontal cortex serotonin induced by PCPA blocked ketamine’s persistent (24 h) antidepressant effect. Serotonin depletion on its own did not affect behavior in the FST. Neither chronic stress nor serotonin depletion individually affected FST immobility 48 h after the final stress session, but the combination of serotonin depletion and restraint stress increased immobility, which was not attenuated by ketamine administered 24 h prior to the FST.
Other studies have reported an increase in serotonin following ketamine exposure. Ketamine (5 or 25 mg/kg SC) dose-dependently increased serotonin in the mPFC of male rats, an effect that was blocked by injecting the AMPAR antagonist NBQX directly into the DRN (Nishitani et al., 2014). Interestingly, when AMPARs were stimulated in the DRN, there was an increase in prefrontal serotonin, yet intra-DRN ketamine did not affect serotonin levels. Instead there was a trend towards reduced PFC serotonin after intra-DRN ketamine. Aside from AMPARs, there seemed to be a connection to cholinergic activity. Intra-DRN injection of an α4β2 nAChR antagonist (DHβE) blocked the ketamine-induced increase in prefrontal serotonin, whereas the intra-DRN injection of the α4β2 nAChR agonist RJR-2403 increased mPFC serotonin, mimicking ketamine.
Antidepressant-like effects in the NSFT were produced in male mice 30 min after ketamine (30 mg/kg IP) or the mGluR2/3 antagonist LY341495; AMPAR antagonism attenuated the effect of both drugs (Fukumoto et al., 2014). Consistent with Gigliucci et al. (2013), serotonin depletion with PCPA blocked the antidepressant-like effect of ketamine and LY341495. Their effects were also blocked by the 5-HT1A antagonist WAY100635, whereas the 5-HT2A/C antagonist ritanserin did not have an effect. Supporting a connection between AMPARs and serotonergic activity, the AMPAR potentiator CX546 mimicked ketamine by reducing latency to feed in the NSFT, which was blocked by serotonin depletion or 5-HT1A antagonism (Fukumoto et al., 2014).
An additional connection between the serotonergic system and AMPAR activity was reported by Yamanaka et al. (2014). Male rhesus monkeys received ketamine (30 mg/kg IV) 100 min before a PET scan and there was a continuous infusion of 7.5 mg/kg/h during the scan. Ketamine increased 5-HT1B receptor binding in multiple regions, particularly in the ventral globus pallidus (ventral GP), NAc, and nucleus reuniens of the thalamus (Tha-Re); this effect was attenuated by AMPAR antagonism. Binding of the SERT ligand DASB was reduced in the NAc, ventral GP, Tha-Re, occipital cortex, and lateral geniculate nucleus (LGN); AMPAR antagonism did not block ketamine’s effect on SERT binding. Although this study did not assess ketamine’s behavioral effects, it suggests one of the consequences of ketamine’s AMPAR agonism may be an elevation of 5-HT1B activity.
Compared to a single dose of the SSRI fluoxetine, ketamine had superior antidepressant effects in BALB/cJ mice (which exhibit a more anxious phenotype at baseline) and its efficacy seemed to rely on intact serotonergic activity (Pham et al., 2017). For systemic administration experiments, ketamine (3 or 10 mg/kg IP) was given 24 h before behavioral testing. In the anxiety-related OFT and EPM tests, neither ketamine nor fluoxetine had anxiolytic-like effects, unlike the positive control diazepam. However, in the FST, NSFT, and splash test (ST), ketamine had prolonged antidepressant effects that were absent with fluoxetine.
During the FST, Pham et al. (2017) used in vivo microdialysis to measure extracellular serotonin. Ketamine did not increase extracellular serotonin in the mPFC or DRN at 3 mg/kg, but it did in the mPFC at 10 mg/kg; therefore, 10 mg/kg was used for the rest of the study. Ketamine (+144%) and fluoxetine (+171%) both increased extracellular serotonin in the mPFC, whereas only fluoxetine elevated serotonin in the DRN. Despite ketamine’s lack of effect on DRN extracellular serotonin, ketamine and fluoxetine were similarly effective at reducing the firing rate of DRN serotonergic neurons.
When serotonin was depleted by three days of tryptophan hydroxylase inhibition with PCPA, which reduced frontal cortex serotonin by ~ 79%, ketamine-induced FST immobility reduction was blocked (Pham et al., 2017). The authors tried to determine the origin of ketamine’s serotonergic effect using local administration. Intra-mPFC ketamine (0.5 μg) decreased immobility in the FST 24 h later and increased extracellular serotonin, whereas intra-mPFC fluoxetine was ineffective; the ketamine-induced increase in serotonin correlated with its antidepressant effect. Intra-DRN administration of the AMPAR antagonist NBQX attenuated intra-mPFC ketamine’s effect on FST behavior and the increase in serotonin, which was only +31% with NBQX versus +157% without.
These results suggest AMPAR activity in the DRN contributes to ketamine’s antidepressant effect and to its elevation of prefrontal serotonin. However, because intra-mPFC ketamine increased prefrontal serotonin and had antidepressant-like effects, it may be acting locally within the PFC. The attenuating effect of AMPAR antagonism in the DRN could be indirect, i.e. AMPAR antagonism in the DRN impedes ketamine’s ability to increase serotonin in the mPFC, a region that receives projections from the DRN, thereby blocking its beneficial effects. Considering systemic fluoxetine causes a similar increase in mPFC serotonin (Pham et al., 2017), it seems unlikely that serotonin elevation itself is sufficient for antidepressant effects.
A few days after ketamine (10 mg/kg IP) administration in CUMS-exposed rats, its antidepressant effects in the FST and SPT were associated with recovery of hippocampal BDNF and p11 expression, whereas knockdown of hippocampal p11 increased FST immobility and reduced sucrose preference in a ketamine-resistant manner (Sun et al., 2016). Ketamine did not affect p11 expression at 30 min, but it did at 72 h. p11 (aka S100A10) is a protein expressed in GABAergic and cholinergic interneurons, as well as monoaminergic, cholinergic, glutamatergic, and GABAergic projection neurons (Svenningsson et al., 2013). Depression-relevant brain regions (e.g. cortex, hippocampus, NAc) express p11, where it interacts with serotonin receptors; p11 overexpression increases 5-HT1B and 5-HT4 receptors.
Depression is associated with reduced p11 in the ACC and ventral striatum (e.g. NAc), and p11 is similarly reduced in genetic animal models of depression. p11 mRNA was reduced in the hippocampus and amygdala of suicide victims, whereas antidepressants (SSRIs, TCAs, MAOIs, ECT) increased expression in the hippocampus and frontal cortex of rodents (Svenningsson et al., 2006, 2013). Overexpression of p11 in mice replicates some of the long-term effects of antidepressant exposure, which is associated with elevated 5-HT1B receptor function; p11 KO mice have a depressive phenotype, reduced 5-HT1B agonist response, and a deficit in response to antidepressants. In contrast, chronic haloperidol, risperidone, and diazepam do not affect p11 expression.
Because of the role p11 plays in mood regulation and the serotonin system, ketamine’s attenuation of stress-induced p11 reduction could be important for its effects and that property would hypothetically be associated with altered serotonergic activity.
Dopamine
Dopamine neuron cell bodies are centered in the midbrain, with neurons in the VTA projecting to corticolimbic regions (e.g. the amygdala, hippocampus, and frontal cortex) and neurons in the substantia nigra (SN) projecting to the dorsal striatum (Cabib and Puglisi-Allegra, 2011). The NAc, which is heavily involved in reward processing, receives dopaminergic projections from the VTA (primarily targeting the NAc shell) and SN (primarily targeting the NAc core). Encountering a stressor can initially increase dopamine release in the NAc, but when that stress persists and cannot be controlled, dopamine release is reduced, which seems to primarily involve neurons originating in the VTA. Elevated dopamine release correlates with attempting to escape the stressor, whereas a reduction in release correlates with helplessness.
There may be an important role of abnormal dopamine transmission in depression, particularly in its associated deficits in cognitive performance, motivation, and the ability to feel pleasure. Research stretching back to the 1970s has shown dopaminergic activity in the NAc is altered by stress (Cabib and Puglisi-Allegra, 2011).
Reports are mixed on this point, but it may have a similar affinity for high-affinity dopamine D2 receptors (Ki = 1.0 μM) as it does for NMDAR, based on research using rat striatal tissue (Kapur and Seeman, 2001, 2002). In cloned D2 receptors expressed in Chinese hamster ovary (CHO) cells, the EC50 for ketamine-induced GTPγS accumulation was only 0.9 μM and it produced partial agonism, with 80% of the effect of dopamine; raclopride attenuated the accumulation and the effect was confirmed to involve D2 receptors since cells without them did not exhibit an increase. By comparison, the EC50 of PCP was 4 μM.
Dopamine receptor agonism and/or dopamine release could conceivably contribute to the psychotomimetic properties of ketamine.
Supporting Research
The effect of ketamine on changes in behavior and dopamine activity caused by the LH paradigm was studied by Belujon and Grace (2014). Male rats were either repeatedly exposed to ketamine (daily for three days) or they received a single dose (5 mg/kg IP) before behavioral testing and neurochemical analysis. The deficit in escape behavior caused by LH exposure was reversed by ketamine, which specifically affected behavior in helpless rats, not in non-helpless rats that were resistant to LH. Helpless rats had a ~50% reduction in VTA dopamine neuron population activity, i.e. the number of spontaneously active dopamine neurons in the VTA. The deficit in dopamine neuron population activity was reversed by ketamine (at 20 min or 2 h) without affecting firing rate or burst firing when compared to the effect of saline in helpless rats, but when compared to control animals, ketamine increased the firing rate and bursting activity in helpless and non-helpless rats.
LH exposure specifically reduced dopamine neuron population activity in the central VTA, without affecting the medial and lateral tracks (Belujon and Grace, 2014). Likewise, when given 20 min before testing, ketamine increased the number of active cells in the center tracks but not the medial or lateral tracks, although it did increase population activity in the medial track at 2 h. Similar effects were found 24 h after exposure, with ketamine attenuating the behavior change seen in helpless rats along with restoring dopamine neuron population activity and firing rate.
Effects in the ventral subiculum of the hippocampus (vSub) and more specifically changes along the vSub-NAc pathway were also studied (Belujon and Grace, 2014). In control animals, high-frequency stimulation (HFS) along that pathway produced LTP, but it caused LTD in helpless rats. When analyzing by NAc region, increased LTD along the vSub to NAc shell pathway was a more specific correlate of helplessness compared to changes along the vSub-NAc core pathway. Ketamine administered 20 min before HFS restored LTP on the vSub-NAc pathway in helpless rats. D1 receptor activity may be implicated in ketamine’s effect since the D1 antagonist SCH23390 eliminated the induction of LTP and restored LTD instead.
Based on the research conducted by Belujon and Grace (2014), it appears ketamine’s reversal of helpless behavior involves the normalization of dopamine neuron activity in the VTA and the restoration of synaptic plasticity along the vSub-NAc pathway, which may depend on D1 receptors. Interestingly, a minority of helpless rats (6/85) did not benefit from ketamine and those same rats did not have a restoration of LTP following ketamine exposure, supporting the importance of vSub-NAc synaptic plasticity.
Li et al. (2015) found the antidepressant-like effect of ketamine (20 mg/kg IP) in male mice in the FST at 30 min was blocked by a nonselective dopamine D2/D3 receptor antagonist (haloperidol), but not by a D1 receptor antagonist (SCH23390). In contrast, a subeffective dose of ketamine (10 mg/kg IP) became effective when combined with a D2/D3 receptor agonist (pramipexole). The same results were observed with MK-801.
In a study on the effects of antidepressant S-ketamine doses on PFC and NAc dopaminergic activity in male mice and rats, S-ketamine increased extracellular dopamine in both regions (Witkin et al., 2016). S-ketamine (17 mg/kg SC) also increased the number of spontaneously active VTA dopamine neurons, an effect that was blocked by NBQX—the change in active cell number was not accompanied by changes in firing rate or bursting activity. Similar results were seen with the mGluR2/3 antagonist LY341495, which increased the number of active dopamine neurons in an NBQX-sensitive manner. In contrast, acute administration of the SSRI citalopram did not alter VTA dopamine neuron activity. Using microdialysis, the authors showed that S-ketamine (10 mg/kg SC) and LY341495 increased extracellular dopamine in the PFC by a max of 300-400%, whereas citalopram did not alter PFC dopamine levels. S-ketamine and LY341495 also increased extracellular dopamine in the NAc (+150-175%). Rats given S-ketamine or LY341495 had a reduction in FST immobility that was comparable to the positive control imipramine; like with the other experiments, citalopram did not acutely affect FST behavior.
Because the rat FST can produce false negatives with SSRIs, NIH Swiss mice were studied (Witkin et al., 2016). In those animals, all three drugs were effective, but citalopram’s impact was smaller than that of ketamine or LY341495. NBQX blocked the effects of S-ketamine and LY341495, but not citalopram. S-ketamine and LY341495 reduced TST immobility in CD-1 mice, but citalopram was not; studies have found the CD-1 strain is less responsive to some antidepressants and exhibits greater depressive-like behavior.
Consistent with the known increase in locomotion produced by drugs that enhance dopaminergic activity, Witkin et al. (2016) observed mild hyperlocomotion with S-ketamine and LY341495, although it was typically nonsignificant. When either drug was coadministered with the dopamine receptor agonist quinpirole, there was a significant potentiation of the locomotor stimulation caused by quinpirole alone, suggesting ketamine can at least potentiate dopaminergic activity. Quinpirole’s effect was not changed by citalopram.
Relevant Research
Humans
NMDAR antagonists like ketamine are thought to elevate extracellular dopamine, at least in some regions (Rabiner, 2007). Subanesthetic doses may reduce binding of raclopride, a selective antagonist of D2 receptors, in the striatum, but the evidence is mixed. Researchers initially observed a reduction in binding that was similar in magnitude to the reduction caused by amphetamine, but multiple subsequent studies did not report any effect. The preclinical literature is also mixed, with some studies showing reduced dopamine receptor ligand binding after ketamine and others showing no effect. Rabiner (2007) notes that human studies typically have not distinguished between binding in the dorsal and ventral striatum, so selective effects in the ventral striatum may be missed. If ketamine impacts the striatal dopaminergic system, it could involve direct dopamine receptor activity or an indirect effect attributable to greater dopamine release.
In a human PET study, seven healthy males received ketamine (0.5 mg/kg IV, 20-min) and its effect on D2 receptor availability was assessed using raclopride as the radioligand (Smith et al., 1998). Ketamine reduced raclopride binding in the striatum, but not in the cerebellum.
Kegeles and colleagues (2000) tested the effect of ketamine on striatal dopamine release induced by amphetamine, as assessed by SPECT using the D2 receptor antagonist IBZM for the radioligand. Eight healthy people received amphetamine (0.25 mg/kg IV) alone or during ketamine infusion (0.2 mg/kg IV bolus, then 0.4 mg/kg/h for 4-hours). Amphetamine’s striatal dopamine release was elevated by ketamine, producing a reduction in IBZM binding. Although both drugs stimulate the cardiovascular system, there was no synergy between them on cardiovascular parameters.
Animals
Ketamine (50 mg/kg IP) and PCP (5 mg/kg IP) had different effects on measures of dopamine activity in rats (Rao et al., 1989). Dopamine activity was assessed by measuring levels of the dopamine metabolite 3-MT. Neither substance affected basal 3-MT levels in the PFC, but when the MAOI pargyline was also used, PCP increased 3-MT, unlike ketamine. Similarly, neither drug affected basal 3-MT levels in the amygdala, but PCP increased 3-MT after pargyline administration. The effects partially changed in the pyriform cortex, where ketamine increased basal 3-MT and both drugs increased 3-MT after pargyline. In the striatum, PCP did not affect basal or post-pargyline 3-MT, whereas ketamine reduced 3-MT under both conditions.
Wang et al. (1995) demonstrated that ketamine (50 mg/kg IP) blocked hypoxia-related basal and potassium-evoked dopamine release in the rat striatum, which occurred without ketamine affecting blood oxygen or CO2.
In rats, tail pinch was found to increase motor activity and striatal dopamine; the dopamine increase was blocked by systemic ketamine (10 mg/kg SC) or MK-801, which did not affect basal dopamine levels on their own (Wheeler et al., 1995). Supporting the involvement of striatal NMDARs, locally applied NMDA increased dopamine, which was partially attenuated by the sodium channel blocker TTX. Intra-striatal MK-801 blocked the increase in striatal dopamine caused by tail pinch. Ketamine was not tested in that part of the experiment, so it is unknown if ketamine has the same effect when applied directly to the striatum.
S-ketamine (0.5 mg/kg IV, 40-min) reduced dopamine D2/D3 receptor binding availability in the striatum, as shown by PET imaging with raclopride in rhesus monkeys (Hashimoto et al., 2016). R-ketamine was ineffective, suggesting S-ketamine may have greater dopaminergic effects that could be mediated by elevated dopamine release.
Ketamine (2, 10, and 50 mg/kg IP) did not affect dopamine release in the NAc core of male mice following electrical stimulation of the VTA (Can et al., 2016). The ketamine enantiomers and some of ketamine’s key metabolites did not produce ≥50% inhibition of dopamine receptors (D1-5) at 10 μM; they also did not have agonist or antagonist activity, as determined by measuring agonist-induced β-arrestin translocation. Ketamine and its enantiomers did not inhibit DAT, NET, or SERT at up to 10 μM.
7.4 S-Ketamine or R-Ketamine?
Introduction
NMDAR antagonism has long been viewed as the primary mechanism of ketamine and that seems to be accurate for its analgesic and anesthetic effects, but it does not clearly apply to ketamine as an antidepressant. Intranasal S-ketamine is the only form of ketamine approved as an antidepressant in the United States, yet many preclinical studies suggest R-ketamine is a more potent (and potentially safer) drug.
Differences between the enantiomers do not seem to involve pharmacokinetics or brain penetration since levels of S-ketamine and R-ketamine are similar in the brain and blood of rodents, and both are cleared from plasma in 4-8 hours (Zanos, 2016; Fukumoto, 2017). The greater potency of S-ketamine for anesthesia and psychotomimetic effects appears to be related to its 4-fold higher NMDAR affinity compared to R-ketamine (Ebert et al., 1997).
The most effective form of ketamine might be the racemate since both enantiomers have antidepressant effects and helpful non-overlapping mechanisms, but if a single enantiomer were to be used, R-ketamine could be preferable. R-ketamine is currently being developed as an antidepressant by Perception Neuroscience under the name arketamine (PCN101). The company claims it has an “improved profile versus both racemic ketamine, as well as esketamine, both in terms of efficacy as well as safety.”
Because there is relatively little research on R-ketamine for depression and no head-to-head trials of the enantiomers, it is too early to say which is more effective. Based on preclinical research in rodents and studies in healthy people, R-ketamine may have a lower side effect burden, although the evidence is mixed. Animal studies have found R-ketamine does not have the same degree of rewarding effects or disruption of sensorimotor gating as S-ketamine (Yang et al., 2015), which may be related to its reduced NMDAR affinity.
S-ketamine (Spravato) is marketed by Janssen Pharmaceutica, which is owned by Johnson & Johnson (J&J). Despite a multidecade history of positive results with intravenous racemic ketamine, relatively unimpressive results were reported by Janssen throughout the S-ketamine development process. For example, Janssen reported mixed results in May 2018 from two of its Phase 3 trials. One study found flexibly-dosed intranasal S-ketamine combined with a newly initiated oral antidepressant produced greater antidepressant effects than only using the oral antidepressant, but that study failed to meet its primary efficacy endpoint, and the second study only showed modest superiority.
After reviewing Janssen’s research, a majority of FDA panelists voted 14-to-2 in favor of approving S-ketamine for depression in February 2019. Janssen had presented results from five Phase 3 trials and a number of others, collectively encompassing 1,700 people. The FDA panel voted in favor of S-ketamine even though just two of its five Phase 3 trials showed a positive result, and in general, S-ketamine only had modest antidepressant effects relative to placebo. Two FDA panelists voted against approval, citing concerns about efficacy and side effects, particularly abuse potential and dissociation.
The panelists who voted to approve S-ketamine cited, among other things, their belief that it would at least help a portion of patients and that patients were comfortable with the side effects (e.g. hallucinations, dissociation) if there was a chance to alleviate their depression. The patient preference data came from a Janssen survey.
S-ketamine nasal spray (Spravato) was officially approved by the FDA in March 2019 for use in conjunction with an oral antidepressant in treatment-resistant patients. Because of its side effects and abuse potential, availability is restricted. Patients must self-administer the drug in the presence of a healthcare provider while in a physician’s office or clinic; S-ketamine cannot be taken home. In discussing the basis for their decision, the FDA noted that Spravato had been tested in three short-term trials and one longer-term maintenance trial in patients initiating a new oral antidepressant. It was only effective in 1/3 short-term trials and in the longer-term trial, where Spravato produced a longer time to relapse compared to patients only receiving an oral antidepressant.
S-Ketamine For Depression
Multiple studies have supported the antidepressant effect of S-ketamine, but because it has generally been studied against placebo, it is difficult to compare the enantiomers using human research. However, the topic has been explored by many animal studies, which have generally shown both enantiomers are effective, but R-ketamine may be more potent and longer-lasting.
Segmiller et al. (2013) reported a reduction in depression in 3/6 TRD patients treated with S-ketamine (0.25 mg/kg IV, 40-min) once or twice per week for four weeks. Patients could continue with stable pharmacotherapy during the trial. Two patients remitted by 120 min, with their HDRS scores falling from 22 to 2 and from 21 to 2 between baseline and 120 min after the last infusion. The first infusion had the greatest effect, while the subsequent infusions maintained its efficacy. Substantial dissociative symptoms occurred in two patients, leading to treatment discontinuation in one.
In an RCT of 30 TRD patients, Singh et al. (2016) studied S-ketamine (0.2 or 0.4 mg/kg IV, 40-min) compared with placebo; nonresponders to placebo received S-ketamine on Day 4. Depression was significantly reduced at 24 h, with no difference between the doses. From baseline to Day 3, the mean reduction in MADRS score was 2.3, 16.3, and 13.4 points with placebo, 0.2 mg/kg, and 0.4 mg/kg, respectively. The response rate (i.e. response on Days 2, 3, or 4) was 0% with placebo, 67% (n=6) with 0.2 mg/kg, and 64% (n=7) with 0.4 mg/kg. Side effects were dose-dependent, with the most common negative effects being dissociation, headache, and nausea. Dissociation did not last more than four hours.
The response rates at 24 h were as follows: 0% with placebo, 67% with 0.2 mg/kg, and 64% with 0.4 mg/kg (Singh et al., 2016). Based on CADSS score, dissociative effects increased with both doses at 40 min, but those effects resolved within four hours; the larger dose seemed to be stronger in this respect. Likewise, psychotomimetic effects were elevated by S-ketamine and 0.4 mg/kg appeared to be stronger than 0.2 mg/kg. The side effects (placebo vs. 0.2 vs. 0.4 mg/kg) were as follows: dissociation (0% vs. 8% vs. 17%); dizziness (0% vs. 8% vs. 3); headache (20% vs. 17% vs. 23%); nausea (20% vs. 25% vs. 10%); vomiting (0% vs. 8% vs. 3%).
Correia-Melo et al. (2017) reported a 48% response rate and 37% remission rate among treatment-resistant MDD (n=23) and BD (n=4) patients one week after treatment, based on a retrospective chart review of patients given S-ketamine (0.25 mg/kg IV, 10-min). The patients had severe baseline depression (MADRS = 36.2) and all were treated with antidepressants; additionally, 20 used atypical antipsychotics and five used lithium. In the first 24 h, 59% had a response and 41% had remission. The most notable side effect was mild to severe dissociation and three patients had psychotomimetic effects during the infusion, specifically with severe dissociative symptoms.
In a Phase 2 DBRCT of 67 TRD patients, twice-weekly intranasal S-ketamine at 28 mg (n=11), 56 mg (n=11), or 84 mg (n=12) was compared with placebo (n=33) during the first part of the trial (Daly et al., 2017). During the second part, 28 placebo-treated patients with moderate-to-severe symptoms were re-randomized. Patients continued their existing antidepressants during the trial. Every S-ketamine dose was superior to placebo on Day 8 and dose-dependence was seen, with MADRS declining by 4.2 points with 28 mg, 6.3 points with 56 mg, and 9.0 points with 84 mg.
The response rates during the first part of the study were as follows (placebo vs. 28 mg vs. 56 mg vs. 84 mg): At 2 h (18% vs. 55% vs. 36% vs. 58%); at 24 h (3% vs. 36% vs. 27.3% vs. 42%); and on Day 8 (6% vs. 9% vs. 18% vs. 42%).
Daly et al. (2017) found the most common side effects (at least 2-fold higher vs. placebo) were dizziness, headache, and dissociation. Most patients treated with S-ketamine experienced a transient increase in blood pressure, with an average peak increase of 19 mmHg for SBP and 10 mmHg for DBP; heart rate also increased by a mean maximum of 9.4 bpm. The perceptual and dissociative effects peaked at 30-40 min and resolved by 2 h; those effects seemed to reduce with repeated dosing. No patient had symptoms indicative of psychosis.
Human Research Favoring S-Ketamine
In two TRD patients treated with both racemic ketamine and S-ketamine, depression was similarly affected by both, but S-ketamine produced less psychotomimetic effects (Paul et al., 2009). The first patient had recurrent major depression and was being treated with mirtazapine, lithium, lorazepam, and zopiclone. They received S-ketamine (0.25 mg/kg IV, 50-min) followed by ketamine (0.5 mg/kg IV, 50-min) the following week. Neither alleviated their depression, but ketamine caused more psychological and perceptual effects, which resolved within one hour.
The second patient had recurrent major depression and was being treated with mirtazapine, lorazepam, amitriptyline, ziprasidone, and zopiclone (Paul et al., 2009). Ketamine (0.5 mg/kg IV, 50-min) was administered followed by S-ketamine (0.25 mg/kg) the following week. On Day 1, their HDRS score fell by 58% with ketamine and 46% with S-ketamine; that effect was largely stable through Day 3 and gone by Day 6. S-ketamine only caused them to feel tired, whereas ketamine produced dizziness, all colors had a “whiff of pink,” and they had a feeling of being “embedded.”
Animal Research Favoring R-Ketamine
R-ketamine seems to be a more potent antidepressant in animal models and produces longer-lasting effects (Zhang et al., 2014; Yang et al., 2015). This suggests NMDAR antagonism is not the key mechanism for the persistent efficacy of ketamine, although it may be comparatively more important for shorter-term effects.
In male mice with depressive behavior caused by the corticosteroid dexamethasone, R-ketamine (10 mg/kg IP) was more potent than S-ketamine (10 mg/kg IP) as an antidepressant and had longer-lasting effects (Zhang et al., 2014). Both enantiomers similarly reduced FST immobility and TST immobility one day after administration, and both attenuated a reduction in sucrose preference two days after administration. But on Day 7, only R-ketamine reduced immobility.
Yang et al. (2015) reported more substantial effects from R-ketamine in multiple rodent assays and it also produced fewer side effects than S-ketamine. Both were effective at improving sucrose preference and TST/FST immobility in the mouse CSDS model, though R-ketamine was more potent. Only R-ketamine was effective in the rat LH model when behavioral effects were tested five days after one 20 mg/kg (IP) dose of either drug. Overall, both had antidepressant and anti-anhedonic effects for at least 6-7 days, but R-ketamine was significantly more potent.
Depressive behavior in CSDS mice correlated with reduced dendritic spine density in the prelimbic (PrL) and infralimbic (IL) regions of the mPFC, as well as in the hippocampal CA3 and DG regions (Yang et al., 2015). Both enantiomers (10 mg/kg IP) attenuated the deficit in those regions eight days after a single dose; R-ketamine was more potent in the DG. Conversely, spine density increased in the NAc core and shell following CSDS exposure and neither enantiomer alleviated that effect.
Pretreatment with either the AMPAR antagonist NBQX or the TrkB antagonist ANA-12 blocked the antidepressant-like effect of both enantiomers (Yang et al., 2015). CSDS reduced BDNF in the PFC, DG, and CA3, but not in the CA1, and it increased BDNF in the NAc. Both enantiomers attenuated the reduction, but they could not reverse the increase in BDNF in the NAc; R-ketamine was more potent in the CA3. Similar effects were seen with TrkB phosphorylation, which was reduced by CSDS in the PFC, CA3, and DG, while it was increased in the NAc. R-ketamine attenuated the deficits and was more effective than S-ketamine in the CA3 and DG, but neither enantiomer was effective in the NAc. CSDS reduced GluR1 protein in the PFC, CA3, and DG, while it increased expression in the NAc; both enantiomers attenuated the effects in every region aside from the NAc.
In terms of side effects, a single dose of S-ketamine (5, 10, or 20 mg/kg) dose-dependently increased locomotion, with a quick return to baseline (Yang et al., 2015). R-ketamine did not affect locomotion. Prepulse inhibition (PPI), which is impaired in schizophrenia, was only reduced by S-ketamine (10 and 20 mg/kg). Their rewarding properties were tested using the conditioned place preference (CPP) test, demonstrating that ketamine (10 mg/kg) increased place preference, as did S-ketamine (5-20 mg/kg), but R-ketamine did not.
Unlike S-ketamine, racemic ketamine and R-ketamine reversed depressive behavior in rats repeatedly treated with corticosterone (Fukumoto et al., 2017). Both enantiomers were active in at least some of the tests and S-ketamine was more potent acutely (at 30 min), but R-ketamine had superior antidepressant effects and a longer duration of efficacy overall. In the mouse FST, S-ketamine was more potent at 30 min and they were equipotent at 24 h. S-ketamine and R-ketamine reduced immobility at 30 min in the TST, with S-ketamine exhibiting greater potency; they were equipotent at 24 h, but only racemic ketamine and R-ketamine were effective at 48 h. Repeated corticosterone exposure increased FST immobility, which was attenuated when either racemic ketamine, R-ketamine, or S-ketamine was given twice (30 min and 24 h before the test), but if only one administration of 10 mg/kg (IP) was given 24 h before the test, the effect was limited to R-ketamine and ketamine. NBQX blocked the antidepressant-like effect of R-ketamine.
The efficacy of ketamine in the corticosterone model of depression may be relevant to TRD in humans since standard antidepressants are less effective in this model and altered stress hormone activity is thought to be involve in depression (Fukumoto et al., 2017).
General Effects of the Enantiomers
In a DBRCT of 60 patients undergoing minor operations, ketamine (n=20) was compared with S-ketamine (n=20) and R-ketamine (n=19) (White et al., 1980). The racemate was given as a 2 mg/kg (IV) induction dose, followed by periodic maintenance boluses of 1 mg/kg; the induction dose was 1 mg/kg for S-ketamine and 3 mg/kg for R-ketamine, while the maintenance dose was 0.5 mg/kg for S-ketamine and 1.5 mg/kg for R-ketamine. S-ketamine was the most potent, requiring an average total dose of 143 mg compared to 346 mg for the racemate and 557 mg for R-ketamine. All groups were similar with regards to heart rate elevation (peak increase of 25-35 bpm for all) and increased SBP (peak increase of 25-32 mmHg). No treatment caused a large change in respiration—there was a peak increase in respiratory rate (RR) of 0.5 for the racemate, 1.2 for S-ketamine, and 0 for R-ketamine.
R-ketamine produced more emergence reactions, with patients reporting vivid dreams, a sense of drunkenness or delirium, and “weird trips” (White et al., 1980). R-ketamine caused less postoperative amnesia, but it was associated with greater restlessness, thrashing, and combative behavior in the recovery room. Compared to S-ketamine, disorientation, agitation, and pain were more prominent with ketamine and R-ketamine. R-ketamine was much less effective as an analgesic, resulting in a greater demand for other analgesics and four times as many patients with pain.
Oddly, while ketamine was an effective anesthetic, a minority of patients reported being conscious during the operation—this was more common with S-ketamine (21% vs. 5% with racemate and 6% with R-ketamine) (White et al., 1980). However, none of the patients were truly aware of what happened during the operation. Each group had a high incidence (83-85%) of dreaming during the operation. Among those who experienced dreaming, 70-80% said the dreams were different from anything they had previously experienced and many reported traveling through space and time while dissociated from their body. Those ‘dream’ states were unpleasant in 22% with R-ketamine, 11% with S-ketamine, and 5% with the racemate, whereas dreaming was considered pleasant in 56% with R-ketamine, 63% with S-ketamine, and 70% with the racemate.
Patient satisfaction was highest among S-ketamine patients, with 85% considering it desirable for anesthesia compared with 65% of patients treated with ketamine and 63% who were treated with R-ketamine (White et al., 1980). Only one S-ketamine patient required adjunctive drugs (e.g. nitrous oxide, thiopental) during the operation because of inadequate anesthesia, which was far superior to 26% with R-ketamine. After the operation, happiness and a sense of wellbeing were more common with S-ketamine. A floating sensation was present in 40-50% of patients.
White and colleagues (1985) also studied the enantiomers in a crossover trial of five healthy males. Each drug was administered via continuous infusion over a 5-7 minute period, specifically with 50 mg/min for ketamine, 25 mg/min for S-ketamine, and 75 mg/min for R-ketamine. Ketamine and its enantiomers produced an anesthetic state for six minutes after the infusion. S-ketamine and ketamine had different EEG effects than R-ketamine, which failed to suppress EEG activity. Each drug produced a similar incidence of dreaming, which was usually considered pleasant. No treatment caused anesthesia and there were no differences in post-anesthesia side effects like dizziness (47%), floating sensation (67%), and diplopia (60%).
In a double-blind crossover study of six males, Oye et al. (1992) studied the impact of the enantiomers on experimentally induced ischemic pain. S-ketamine was 4x as potent for inducing a subjective sensation of being on a drug and there was a similar potency difference for analgesia. At equianalgesic doses, S-ketamine produced a greater reduction in alertness and more somatosensory disturbances (e.g. a feeling of detachment or of floating in the air).
Similarly, in a study of 16 female patients with acute pain after oral surgery and seven female patients with chronic neuropathic orofacial pain, the analgesic potency of S-ketamine was 4x higher than R-ketamine (Mathisen et al., 1995). The enantiomers had similar mental effects, but relative to their analgesic effects, S-ketamine had more disturbing side effects than R-ketamine:
- Side effects for S-ketamine (0.45 mg/kg IM) (n=9): 100% blurred vision, 78% altered hearing, 89% dizziness, 100% proprioceptive disturbances, 56% illusion, 0% for sedation, dreams, or hallucinations
- Side effects for R-ketamine (1.8 mg/kg IM) (n=9): 78% blurred vision, 67% altered hearing, 89% dizziness, 56% proprioceptive disturbances, 22% illusions, and 0% for sedation, dreams, or hallucinations.
- Side effects for racemic ketamine (0.9 mg/kg IM) (n=7): 85% blurred vision, 57% altered hearing, 100% dizziness, 71% proprioceptive disturbances, 57% illusions, 0% sedation, 43% dreams, 43% hallucinations
Vollenweider et al. (1997) found S-ketamine (15 mg/kg IV) caused psychotic-like effects, but R-ketamine (15 mg/kg) did not; they studied the enantiomers in 10 healthy males. As shown by PET imaging, S-ketamine increased glucose utilization in the frontal cortex (ACC, parietal, and left sensorimotor cortex) and in the thalamus, whereas R-ketamine reduced glucose utilization in multiple areas, including the left insula and temporomedial cortex. The increase in frontal and left temporal cortex glucose utilization induced by S-ketamine correlated with its ego-disintegration and hallucinatory effects.
S-ketamine (84 mg intranasal) did not impair driving performance 8 h after administration in a study of 26 healthy people who completed a standardized on-road driving test (van de Loo, 2017). In contrast, mirtazapine increased standardized deviation of lateral position (SDLP, i.e. weaving).
Morrison et al. (2018) compared S-ketamine (84 mg intranasal) with placebo in a DBRCT crossover study of 24 healthy people. S-ketamine impaired cognitive performance at 40 min, but there was no impairment at 2, 4, or 6 h. It also increased scores on the Mental Effort Scale, indicating S-ketamine made it more subjectively difficult to complete the tests; that effect resolved within two hours. The acute side effects of S-ketamine versus placebo were as follows: dizziness (67% vs. 4%); headache (21% vs. 13%); disturbances in attention (29% vs. 0%); somnolence (25% vs. 4%); dysgeusia (13% vs. 4%); hypoesthesia (17% vs. 0%); paresthesia (13% vs. 4%); fatigue (29% vs. 0%); feeling abnormal (25% vs. 0%); feeling drunk (17% vs. 0%); feeling hot (17% vs. 0%); nausea (38% vs. 0%); vomiting (21% vs. 0%); blurred vision (17% vs. 0%); and visual hallucination (13% vs. 0%).
7.5 BDNF-TrkB Signaling
Background
Nerve growth factor (NGF), the first neurotrophin, was discovered in the 1950s and in the decades since that discovery, a number of neurotrophins have been identified, including BDNF and neurotrophins 3, 4, 5, and 6 (Teixeira et al., 2010). Of those, BDNF is the most widely distributed in the CNS, where it regulates the growth and maintenance of axons and dendrites, affects neurotransmitter release, and participates in LTP. Through these effects, BDNF is an important regulator of synaptic plasticity. It is known to be involved in the development and survival of dopaminergic, GABAergic, cholinergic, and serotonergic neurons (Autry and Monteggia, 2012).
Impaired BDNF activity in adults is not fatal (unlike early in development), but it may contribute to psychiatric and neurological disorders (Teixeira et al., 2010). BDNF disruption can produce neurodegeneration associated with deficits in dendritic spine number and arborization, neural atrophy, and impaired neurogenesis. Those changes appear to be associated with depression and cognitive impairment. However, BDNF itself does not control mood and there is an absence of evidence clearly demonstrating that the mere reduction of BDNF signaling causes depressive behavior on its own.
BDNF is synthesized following the production of a precursor, prepro-BDNF, which is cleaved into pro-BDNF; subsequently, pro-BDNF is cleaved into mature BDNF (Autry and Monteggia, 2012). Mature BDNF primarily signals through the high-affinity TrkB receptor, while pro-BDNF has different effects associated with signaling through the low-affinity neurotrophin receptor p75, which appears to have a role in apoptosis. Binding of BDNF to TrkB causes receptor dimerization and autophosphorylation of tyrosine residues, which yields docking sites for intracellular signaling cascades like Ras/MAPK, PI3K/Akt, and PLC. Through those signaling pathways, BDNF supports neuronal differentiation and survival. Additionally, BDNF-TrkB binding can affect NMDAR currents.
Activation of the PLC γ pathway results in PKC activation, while the PI3K pathway activates Akt (PKB), and the MAPK pathway activates multiple effectors (Autry and Monteggia, 2012). The longer-lasting effects of BDNF primarily occur via transcription regulation; those effects are downstream of the PI3K and MAPK pathways. BDNF and TrkB are localized at pre- and postsynaptic sites. Presynaptic signaling regulates neurotransmitter release, whereas postsynaptic signaling alters the function of ion channels, including AMPARs, NMDARs, sodium channels, potassium channels, and transient receptor potential channels (TRP channels).
Peripheral BDNF is stored in platelets and can be released upon stimulation (Teixeira et al., 2010). In plasma, the concentration of BDNF is in the pg/mL range, but it is in the ng/mL range in serum; because of the difference in concentration, studying peripheral serum is preferable to plasma. For practical reasons, human studies typically examine peripheral BDNF levels. This can pose problems since peripheral neurotrophins and other molecules do not necessarily correlate with levels in the CNS. Rodent research has also shown there are appreciable levels of NGF, BDNF, and NT-3 in peripheral tissues (e.g. heart, liver, lung), making it harder to associate peripheral changes with effects in the CNS (Lommatzsch et al., 2005).
While not ideal, peripheral BDNF seems to be an acceptable indicator of BDNF in the CNS; for example, there was a strong correlation between plasma and CSF BDNF in first-episode psychosis patients (n=34) (Pillai et al., 2010). Rodent research also indicates peripheral BDNF can readily enter the brain. Pan et al. (1998) intravenously administered BDNF to mice and demonstrated rapid influx into the brain; efflux to the periphery was also shown using ICV administered BDNF.
Ketamine’s Effect on Neurotrophins
There is an abundance of evidence supporting a role for neurotrophin activity in the antidepressant and synaptogenic effects of ketamine. Ketamine is consistently associated with enhanced synaptic plasticity in regions like the hippocampus and PFC, and this correlates with elevated BDNF and greater BDNF-TrkB signaling.
Supporting Research
Animals
Ketamine’s acute antidepressant-like effect in the FST is associated with increased hippocampal BDNF in male rats (Garcia et al., 2008). Although both ketamine (10 or 15 mg/kg IP) and imipramine were effective in the FST at 1 h, only ketamine increased hippocampal BDNF; the increase was exclusively seen with the 15 mg/kg dose.
Autry et al. (2011) found the rapid antidepressant effect of ketamine (3 mg/kg IP) was dependent on BDNF synthesis, with inducible BDNF KO mice failing to show an effect in the FST. Ketamine and MK-801 reduced FST immobility at 30 min in control mice, but not in BDNF KO mice. There was a rapid but transient increase in hippocampal BDNF expression since protein levels were elevated at 30 min, but not at 24 h; in contrast, ketamine did not affect BDNF mRNA expression at either timepoint. Similar effects were observed with proBDNF. This suggests ketamine quickly increases BDNF protein translation in the hippocampus and that increase is at least relevant to its acute behavioral effects.
Mice with the BDNF Val66Met polymorphism exhibited atrophy of distal apical dendrites and reduced dendritic spine density and diameter at baseline (Liu et al., 2012). Met/Met mice had a greatly reduced synaptogenic response to ketamine (10 mg/kg IP) and they did not have an antidepressant-like effect in the FST at 24 h. Both Val/Met and Met/Met had impaired serotonin- and orexin-induced EPSCs in layer V pyramidal cells in the mPFC, consistent with impaired synaptic potentiation.
Hippocampal BDNF expression and mTOR activity were increased by ketamine at antidepressant doses in male rats (Yang et al., 2013). FST immobility at 30 min was dose-dependently reduced by ketamine (5-15 mg/kg IP). 10 and 15 mg/kg increased hippocampal BDNF, while every dose increased p-mTOR.
Lepack et al. (2014) observed that BDNF release is dependent on AMPAR activity and L-type VDCCs, and intact functioning at those sites is necessary for ketamine’s antidepressant effect in male rats. The role of BDNF was tested by infusing an anti-BDNF antibody. While ketamine on its own reduced FST immobility at 24 h, that was fully blocked by the antibody; the antibody by itself did not affect behavior. When rats were pretreated with VDCC antagonists (nifedipine or verapamil), ketamine’s antidepressant-like effect in the FST was attenuated; the VDCC blockers did not affect behavior on their own. In vitro, ketamine (0.5 μM) was applied to cortical neuron cultures for 15 min, 60 min, or 6 h. Every exposure duration increased BDNF by ~50%. Pretreatment with either NBQX or verapamil blocked ketamine-induced BDNF release, supporting a role for AMPARs and VDCCs.
Yang and colleagues (2015) reported that the TrkB inhibitor ANA-12 blocked the antidepressant-like effects of R-ketamine (10 mg/kg IP) and S-ketamine (10 mg/kg IP) in CSDS-exposed mice at 24 h.
Ketamine attenuated depressive behavior for at least one week in the mouse CSDS model and attenuated many of the CSDS-induced changes in synaptic protein expression (Zhang et al., 2015). TST and FST immobility were improved in male CSDS-susceptible mice by ketamine (10 mg/kg IP), the TrkB antagonist ANA-12, and the TrkB agonist 7,8-DHF. Every treatment increased sucrose preference on Days 1 and 3, but ketamine continued to improve sucrose preference until at least Day 7. No drug had antidepressant-like effects in control (non-CSDS) mice. CSDS increased proBDNF exclusively in the PFC, which was attenuated by ketamine. BDNF was reduced by CSDS in the PFC, DG, and CA3, but not in the CA1, whereas it increased BDNF in the NAc. Ketamine attenuated every change in BDNF except for the increase in the NAc. 7,8-DHF and ANA-12 had no effect in any region.
A similar pattern was reported for the synaptic proteins PSD95 and GluR1. CSDS reduced PSD95 in the PFC, DG, and CA3, while increasing it in the NAc (Zhang et al., 2015). Ketamine and 7,8-DHF attenuated the reduction in the PFC, DG, and CA3, but not the increase in the NAc. ANA-12 only attenuated the increase in the NAc. CSDS reduced GluR1 in the PFC, DG, and CA3, while increasing it in the NAc. Ketamine and 7,8-DHF attenuated every change except for the increase in the NAc, whereas ANA-12 exclusively attenuated the increase in the NAc.
There was no difference in proBDNF with any treatment or stress condition eight days after drug exposure, but stress and ketamine continued to affect BDNF (Zhang et al., 2015). CSDS reduced BDNF in the PFC, DG, and CA3, while increasing it in the NAc; this was attenuated by ketamine, except for NAc change. Neither 7,8-DHF nor ANA-12 had an effect. CSDS reduced PSD95 in the same regions, along with increasing it in the NAc; ketamine attenuated every change other than the increase in the NAc. Again, ANA-12 and 7,8-DHF were ineffective. CSDS also reduced GluR1 in the PFC and DG, but not in the CA1 or CA3, and it increased GluR1 in the NAc. Ketamine elevated GluR1 in the PFC, DG, and CA3. In contrast, neither 7,8-DHF nor ANA-12 had any effect.
Ketamine and the mGluR2/3 antagonist MGS0039 attenuated the increase in TST (24 h) and FST (48 h) immobility observed in male CSDS-susceptible mice; both drugs also improved sucrose preference (Dong et al., 2017). CSDS reduced BDNF in the PFC, DG, and CA3, but not the CA1, and it increased BDNF in the NAc. MGS0039 attenuated the change in the PFC and DG, while ketamine was effective in the PFC, DG, and CA3; neither drug reversed the elevation in NAc BDNF.
Phosphorylation of TrkB was similarly altered by CSDS in the same regions and MGS0039 attenuated a reduction in the CA3, while ketamine was effective in the PFC, DG, and CA3; neither drug affected the increase in p-TrkB in the NAc (Dong et al., 2017). CSDS also affected GluR1 in these regions, with MGS0039 attenuating a reduction in the PFC and DG, while ketamine was effective in the PFC, DG, and CA3, but neither drug affected the increase in GluR1 in the NAc. Likewise, PSD-95 was altered with the same pattern and MGS0039 reversed those changes in the PFC and DG, while ketamine was effective in the PFC, DG, and CA3, but neither drug was effective in the NAc.
CSDS reduced dendritic spine density in the PrL region of the mPFC (but not the IL region), as well as in the hippocampal DG and CA3 regions (Dong et al., 2017). Ketamine and MGS0039 attenuated those reductions. CSDS had the opposite effect in the NAc, increasing dendritic spine density in the NAc core and shell; neither drug reversed this effect.
In male rats, the alleviation of CUS-induced behavioral and neurochemical effects was associated with altered p-CREB, BDNF, GLT-1, and PSD95 in the hippocampus (Liu et al., 2016). Ketamine (10 mg/kg IP) reversed a CUS-induced increase in TST immobility at 24 h, an effect blocked by the TrkB antagonist K252a. In the hippocampus, CUS decreased the Bcl2/Bax ratio, BDNF, p-CREB, and GLT-1. Those changes were reversed by ketamine, but its efficacy was blocked by K252a. Additionally, PSD95 was reduced by CUS and ketamine alleviated that change in a K252a-sensitive manner.
CUS reduced the density of GFAP+ cells (i.e. astrocytes), but not NeuN+ cells (i.e. neurons), in the DG, CA1, and CA3 regions of the hippocampus (Liu et al., 2016). Treatment with ketamine, K252a, or both failed to reverse the reduction in GFAP+ cells. In the same regions, CUS increased cleaved caspase 3-positive cells, a marker of apoptosis, with nearly all cleaved caspase-3 colocalized with GFAP instead of NeuN, suggesting the loss of astrocyte density was associated with increased apoptotic signaling. Ketamine attenuated the CUS-induced increase in hippocampal apoptosis, an effect reversed by K252a. The authors also studied neuron morphology in the CA1 region. CUS reduced dendritic branch number, dendrite length, and spine density. Ketamine attenuated the reduction in spine density; that effect was blocked by K252a.
Different effects on BDNF mRNA and protein expression were observed in two mouse strains, namely male Kunming (KM) mice and male ICR mouse (Xue et al., 2016). In KM mice, ketamine (30 mg/kg IP) reduced TST immobility at 30 min, 3 h, Day 1, Day 2, Day 3, and Day 5, whereas immobility was only reduced at 30 min, 3 h, and Day 1 when ICR mice received ketamine (50 mg/kg IP). Ketamine increased BDNF mRNA at 3 h, Day 1, and Day 2 in KM mice, but only at 3 h in ICR mice. Strain differences were also present in ketamine’s effect on hippocampal CREB. Ketamine did not alter total CREB or p-CREB on Days 1 or 2 in ICR mice, but p-CREB and total CREB were increased on Day 1 in KM mice; that effect was lost by Day 2.
Because the increase in CREB protein could be related to transcription upregulation, the authors tested the effect of ketamine on CREB gene expression in the hippocampus (Xue et al., 2016). ICR mice showed no change in CREB mRNA at 30 min or Day 1; in contrast, KM mice had an increase at 3 h, but not at 30 min or Day 1. PKA expression was increased one day after ketamine in KM mice, but not in ICR mice. However, the PKA inhibitor H89 failed to attenuate ketamine’s effect in KM mice and it did not block the ketamine-induced increase in BDNF mRNA. A role for mTOR signaling was supported since rapamycin blocked ketamine’s antidepressant effect and reversed its upregulation of BDNF mRNA. This suggests that ketamine depends on the mTOR pathway and BDNF activity, but not the PKA/CREB/BDNF pathway.
Ma et al. (2017) studied the effect of ketamine in the adult hippocampus of mice. Within 24 h, ketamine (7 mg/kg IP) accelerated doublecortin-positive (DCX+) progenitor differentiation into mature neurons, with its largest effect in the DG. That dose did not affect normal neural progenitor cell (NPC) proliferation and differentiation in the hippocampus, as determined by Ki67+ and DCX+ cells at 24 h, but one week after ketamine there was an increase in DCX+ progenitor cell number. At both 24 h and one week, ketamine increased the generation of newborn neurons (NeuN+, DCX-) among BrdU+ cells.
The functional status of ketamine-induced newborn neurons was tested using whole-cell patch clamp recordings (Ma et al., 2017). These neurons had normal dendritic branching and normal electrophysiological activity. Because normal spontaneous postsynaptic currents (sPSCs) were recorded, the newborn neurons appeared to have been integrated into the local neural circuitry and functional synapses were formed. To determine the importance of accelerated DCX+ to NeuN+ cell differentiation for the behavioral effects of ketamine, Nestin+ neural stem cells and their progeny were eliminated via genetic manipulation. This blocked ketamine’s antidepressant effect in the FST and NSFT, indicating ketamine is dependent on increased differentiation of late-stage progenitor cells into mature neurons.
A role for progenitor cell differentiation was further supported using the DNA alkylating agent temozolomide (TMZ), which targets BrdU-incorporating cells (Ma et al., 2017). Although both control and TMZ-exposed mice had accelerated newborn neuron generation post-ketamine, that effect was reduced by 80% in the TMZ-exposed mice. Those mice did not receive an antidepressant-like effect from ketamine in the FST. Importantly, the ketamine-induced acceleration of DCX+ progenitor differentiation occurred concurrently with an increase in BDNF in the DG; therefore, the role of BDNF-TrkB signaling was assessed. TrkB elimination did not affect basal behavior, but it blocked ketamine’s effect in the FST and NSFT. The generation of newborn neurons post-ketamine was also impaired in mice with TrkB-deleted NPCs and those mice had greater immobility in the FST 24 h and one week after ketamine compared to normal ketamine-treated animals. However, ketamine still produced an acute (1 h) antidepressant effect in mutant mice, suggesting that ketamine’s persistent efficacy depends on neurogenesis, but not its acute effect.
Because TrkB connects with a variety of effector pathways, Ma et al. (2017) attempted to determine which was driving the effects. They examined the ERK (RAS/MAPK) pathway using the selective MEK inhibitor SL3232736 and found that it attenuated the ketamine-induced increase in p-ERK in the DG, blocked ketamine’s antidepressant-like effect at 24 h, and attenuated its neurogenic effect.
In Vitro
Lepack et al. (2016) studied ketamine and other putative antidepressants in rat primary neuronal cultures. Ketamine, like GLYX-13 and the mGluR antagonist LY341495, increased phosphorylation of ERK and p70S6K, as well as BDNF release. The increase in p-ERK occurred at ketamine concentrations of 10 to 500 nM, whereas larger concentrations (1 and 10 μM) were ineffective; therefore, 100 and 500 nM were used for subsequent experiments. p-ERK increased within 15 min and remained elevated for at least 24 h. p-mTOR was found at inconsistent levels, but p-p70S6K was more reliably elevated by ketamine, indicative of mTORC1 activation. Ketamine also increased p-ERK and p-p70S6K in hippocampal cultures.
GLYX-13 had a ketamine-like inverted U dose-response curve for p-ERK elevation, with the largest concentrations showing no effect, while smaller amounts increased p-ERK (Lepack et al., 2016). Unlike ketamine, GLYX-13, and LY341495, neither fluoxetine nor desipramine acutely affected these measures. AMPAR antagonism with NBQX blocked the ketamine-induced increase in p-ERK and it was also effective against GLYX-13 and LY341495. To test the role of BDNF-TrkB signaling, the TrkB inhibitor K252a was used, which blocked the p-ERK increase from all three drugs. Therefore, p-ERK elevation is dependent on AMPAR activation and BDNF-TrkB signaling.
At 1 h, ketamine increased BDNF release, as did GLYX-13 and LY341495, whereas acute fluoxetine and desipramine did not affect BDNF release (Lepack et al., 2016). NBQX attenuated the increase in BDNF release. Because AMPAR activity seemed to be required, which is consistent with a role for glutamate release and its resultant excitatory effect, the effect of GABAA receptors was evaluated. The GABAA antagonist bicuculline stimulated p-ERK on its own, whereas cells treated with the GABAA agonist muscimol had an attenuation of p-ERK elevation and BDNF release from ketamine, GLYX-13, and LY341495. This supports the hypothesis that ketamine reduces inhibitory output from GABAergic interneurons, thereby disinhibiting pyramidal neurons. Ketamine increased dendrite complexity at 24 h, with an increase in dendritic branch crossings 50 and 100 μm from the soma; similar effects occurred with GLYX-13 and LY341495.
Opposing Research
Although chronic (14-day) ketamine at 5-15 mg/kg (IP) in male rats has antidepressant effects in the FST, it does not persistently increase BDNF, unlike with acute administration (Garcia et al., 2008). Both chronic imipramine and ketamine reduced FST immobility at 60 min. Instead of becoming weaker, when a subeffective dose (5 mg/kg) is chronically administered, it can become effective. Neither chronic ketamine nor imipramine affected hippocampal BDNF at any dose.
In rats undergoing a CMS procedure, acute or chronic (7-day) ketamine (15 mg/kg IP) alleviated stress-induced behavioral and biochemical changes in male rats, although it appeared to be more effective when given chronically (Garcia et al., 2009). The ketamine-induced effects occurred without any change in hippocampal BDNF from ketamine or stress. Chronic ketamine reversed the deficit in sweet food consumption caused by CMS, but a single dose was ineffective. The interpretation of these results is complicated by an increase in sweet food intake among non-stressed rats who received chronic ketamine, suggesting a nonspecific change in eating behavior could also be involved.
CMS increased serum corticosterone and adrenocorticotropic hormone (ACTH) levels, which were reversed by acute and chronic ketamine (Garcia et al., 2009). Similarly, adrenal gland weight increased due to stress exposure and ketamine (acute and chronic) attenuated that effect. During 40 days of observation, CMS rats did not gain weight, but they started to gain weight after acute or chronic ketamine treatment; instead of being related to antidepressant effects, this could just mean NMDARs are involved in the regulation of eating during stress. Stress did not affect hippocampal BDNF, nor did acute or chronic ketamine, both in stressed and nonstressed rats. A potential explanation for this result is that hippocampal samples were taken one week after the end of the chronic stress procedure, the results may have been different if BDNF was assessed closer to the stress procedure.
Lindholm et al. (2012) reported that heterozygous BDNF KO male mice have an attenuated antidepressant effect with imipramine in the FST, but ketamine and the AMPAR potentiator LY451646 were unaffected. Ketamine (20 or 50 mg/kg IP) was effective in the FST acutely (45 min), but both ketamine and LY451646 were ineffective one week later. Neither drug affected BDNF levels or TrkB phosphorylation in the hippocampus at 45 min or one week. Heterozygous BDNF KO mice had a ~60% reduction in BDNF; therefore, the lack of impact may indicate ketamine is less reliant on BDNF-TrkB activity than is commonly believed. Alternatively, the remaining amount of BDNF could be sufficient to permit ketamine’s effect. Another factor is that BDNF was assessed after a 50 mg/kg dose and multiple studies have found higher doses of ketamine are less effective or ineffective compared lower amounts. The BDNF deficit in heterozygous BDNF KO mice did not affect basal FST behavior, but it blunted the effect of imipramine.
7.6 GSK-3
Glycogen synthase kinase-3 (GSK-3), which exists as two isoforms (GSK-3α and GSK-3β), is a prolific kinase that phosphorylates more than 100 proteins and is present in the cytosol, mitochondria, nucleus, and other subcellular compartments (Beurel et al., 2015). Originally identified for its role in insulin receptor signaling, it is now known to influence many cellular activities, including through regulation of gene expression via targeting transcription factors like CREB, heat shock factor 1 (HSF1), NF-kB, STAT3, and NFAT. Among other things, regulation of those pathways affects neural survival and growth, which appear to be components of ketamine’s mechanism.
GSK-3 is primarily regulated by serine phosphorylation (Ser21 on GSK-3α and Ser9 on GSK-3β), which inhibits its activity. One way inhibitory phosphorylation is increased is through activation of the PI3K/Akt pathway (Beurel et al., 2015; Freland and Beaulieu, 2012). It is positively regulated through tyrosine phosphorylation (Tyr279 on GSK-3α and Tyr216 on GSK-3β).
Multiple psychiatric disorders have a hypothetical connection to the Akt/GSK-3 pathway (Freland and Beaulieu, 2012).
A Potential Role for Lithium
Lithium, an alkali metal that is usually delivered as a carbonate or citrate salt, entered medical use in the 1800s and became a common treatment for mania in the mid-1900s (Freland and Beaulieu, 2012). An early observation that contributed to the recognition of GSK-3 as an important protein in psychiatry was the identification of GSK-3 as a target of lithium, which is widely used for bipolar disorder. Through its competition for a magnesium binding site on GSK-3, lithium directly inhibits its activity, and this may be important for its mood-stabilizing effects (Ryves and Harwood, 2001). Aside from competition with magnesium, lithium increases inhibitory phosphorylation of GSK-3, a property shared by some antidepressants. When that regulatory action is blocked via a serine-to-alanine mutation, rodents exhibit greater amphetamine-induced locomotor activation and they are more susceptible to stress-induced depressive behavior, implicating GSK-3 in manic-type disorders and in depression.
Much of lithium’s effect on GSK-3 in humans likely comes through an indirect route since its therapeutic serum concentration (0.5-1.2 mM) is well below its affinity for GSK-3, where it has a Ki of 3.5 mM for GSK-3α and 2 mM for GSK-3β (Freland and Beaulieu, 2012). At human-relevant concentrations, acute and chronic lithium activates Akt in the frontal cortex, hippocampus, and striatum of rodents. Activation of Akt could be a major indirect contributor to lithium’s GSK-3 inhibition, as Akt negatively regulates GSK-3.
Ketamine has multiple mechanistic overlap points with lithium, including mTORC1 activation and enhanced BDNF activity. Mice chronically treated with lithium showed mTOR activation in renal collecting ducts and rapamycin reversed lithium-induced medullary cell proliferation, although it did not prevent an increase in medullary levels of p-GSK-3β (Gao et al., 2013). In vitro, blocking the Akt pathway, such as by inhibiting PI3K, attenuates inhibitory phosphorylation of GSK-3α and GSK-3β (Cross et al., 1995). Akt is an important downstream effector of PI3K that regulates mTORC1. In cultured rat cortical neurons, a therapeutic concentration of lithium increased BDNF mRNA and the activity of BDNF promoter IV, which contributes to BDNF transcription (Yasuda et al., 2007). Lithium’s activation of promoter IV was mimicked by pharmacological inhibition of GSK-3 and by gene silencing of GSK-3α or GSK-3β.
Separate from its general efficacy in bipolar disorder, lithium may have a specific anti-suicidal effect that could hypothetically combine well with the anti-suicidal effect of ketamine. In a review of 32 RCTs covering 1,400 patients treated with lithium compared with 2,100 patients treated with other drugs, lithium was associated with a positive effect on suicide and self-harm risk in the context of mood disorders, including BD, unipolar depression, and dysthymia (Cipriani et al., 2005).
Supporting Research
Humans
In a DBRCT crossover study of treatment-resistant BD patients (n=36) who had not adequately responded to valproate or lithium, ketamine had an anti-anhedonic effect and there was a trend-level superior improvement in patients maintained on lithium compared to patients maintained on valproate (Lally et al., 2014). This provides weak evidence for either a negative effect of valproate or an additive effect of lithium.
Animals
Ketamine increased inhibitory serine phosphorylation of GSK-3α and GSK-3β in the cerebral cortex and hippocampus of mice, and its antidepressant-like effects in animals exposed to the LH paradigm were blocked by serine-to-alanine mutation, which caused GSK-3 to be maximally active (Beurel et al., 2011). A larger acute dose of lithium than would be used in typical therapeutic scenarios also mimicked the antidepressant effects of ketamine.
A typically inactive dose of ketamine in rats (1 mg/kg IP) was potentiated by lithium chloride (10 mg/kg IP) and by SB216763, which preferentially inhibits GSK-3β (Liu et al., 2013). Lithium enhanced GSK-3β phosphorylation, mTORC1 activation, increased synaptic spine density and diameter, increased EPSCs in mPFC layer V pyramidal neurons, and antidepressant-like effects in the FST for up to one week after administration, occurring alongside a sustained increase in the number and function of excitatory synapses. This supports a role for GSK-3 inhibition in ketamine’s antidepressant and synaptogenic effects, and it suggests that ketamine’s overlapping mechanisms with lithium could be exploited for therapeutic benefit.
Consistent with the findings of Beurel et al. (2011), an ineffective dose of ketamine (2.5 mg/kg IP) in mice exposed to chronic restraint stress became effective in the FST when it was preceded by three weeks of subtherapeutic lithium (Chiu et al., 2015). Continued administration of lithium also sustained the antidepressant-like effects and restoration of mPFC dendritic spine density caused by ketamine (50 mg/kg IP) for at least two weeks, longer than the effect of ketamine alone. Subtherapeutic lithium pretreatment combined with a single subtherapeutic dose of ketamine mimicked important effects of therapeutic ketamine doses, such as increased phosphorylation of TrkB, Akt, ERK, and GSK-3β in the PFC; in contrast, chronic restraint stress decreased GSK-3β phosphorylation in the PFC.
Chiu and colleagues (2015) also reported an increase in oxidative stress with acute ketamine (50 mg/kg), as shown by elevated lipid peroxidation, catalase activity, and oxidized glutathione in the PFC, hippocampus, and striatum of stressed mice. Lithium attenuated those increases, suggesting it may improve both ketamine’s efficacy and safety.
Opposing Research
Although ketamine (10 mg/kg IP) has acute and prolonged antidepressant effects in CMS-exposed male mice, the GSK-3β inhibitor SB216763 did not improve FST or TST immobility at 48 h (Ma et al., 2013). SB216763 also failed to alleviate the anhedonic-like effect of chronic stress in the SPT, whereas ketamine improved sucrose preference for at least eight days after administration. Comparing ketamine with a GSK-3β inhibitor is a poor way to determine the role of GSK-3 inhibition in ketamine’s activity, but this at least suggests that GSK-3β inhibition, while it may contribute to the effect of ketamine, is not sufficient to improve depressive behavior.
7.7 Modulation of Inflammation and Immune System Activity
Supporting Studies
Humans
Ketamine produced antidepressant effects in unmedicated MDD patients (n=16) along with an anti-inflammatory effect that correlated with changes in mood (Yang et al., 2015). There was a 75% response rate following ketamine (0.5 mg/kg IV, 40-min) administration, resulting in 12 responders and 4 nonresponders. Compared to control subjects (n=24), baseline serum IL-1β and TNF-α were higher in patients overall, but baseline IL-1β and IL-6 were also higher in responders than in nonresponders, with IL-6 showing no difference between the control and nonresponder groups.
After treatment, serum IL-1β was reduced at 230 min and Day 1, and IL-6 was reduced from 230 min to three days after infusion in responders (Yang et al., 2015). In contrast, those changes were not seen in nonresponders. Serum TNF-α and kynurenine/tryptophan ratio were not affected in either group. This suggests baseline cytokine status, particularly IL-6, could be a predictor of response and cytokine modulation by ketamine may be important for its antidepressant effect. However, the very small number of nonresponders limits the generalizability of these results.
An analysis of data from three studies of 80 inpatients with treatment-resistant MDD (n=49) or BD (n=31) found correlations between ketamine’s effect and plasma adipokine levels (Machado-Vieira et al., 2017). Ketamine (0.5 mg/kg IV, 40-min) was given as a single infusion and all patients were unmedicated except for 15 BD patients who were using lithium or valproate. Adipokines (e.g. leptin, adiponectin, TNF-α) are cytokines secreted by adipocytes (fat cells). Patients with lower baseline adiponectin had a superior response to ketamine at 230 min and Day 1, whereas baseline leptin and resistin did not correlate with response. Ketamine reduced leptin at 230 min, but that change was gone by Day 1. Resistin was reduced at 230 min and further reduced on Day 1.
When patients were separated by depression subtype, other correlations were found (Machado-Vieira et al., 2017). People with ‘atypical’ depression had higher leptin levels than those who were not classified as atypical or melancholic, and resistin was higher in the ‘atypical’ group compared to melancholic patients. The difference in leptin levels was lost after controlling for BMI, which was significantly correlated with baseline leptin but not baseline adiponectin or resistin. BMI also independently correlated with antidepressant response at 230 min and Day 1, with a superior response in patients with higher BMIs. BD patients had lower plasma resistin than MDD patients throughout the trial, with the greatest difference on Day 1; neither adiponectin nor leptin differed between the groups and there was no effect of lithium versus valproate use.
Kadriu et al. (2018) studied a hypothetical connection between the RANKL/RANK/OPG system—which is associated with bone remodeling and inflammation—and ketamine’s effect. Unmedicated TRD patients (n=28) had a smaller baseline OPG/RANKL ratio and lower plasma osteopontin (OPN) compared with healthy controls (n=16). Ketamine (0.5 mg/kg IV, 40-min) increased both in TRD patients, while reducing RANKL; ketamine did not affect bone markers in control subjects. The increase in OPG/RANKL ratio was significant at 230 min and there were nonsignificant trends on Days 1 and 3, while the increase in OPN was significant on Days 1 and 3. Patients with lower OPN/RANKL ratio at baseline, which may be indicative of a bone loss state, experienced an increase at 230 min, Day 1, and Day 3. RANKL was similar between groups at baseline, but the patients had a decrease at 230 min and Day 3, whereas those changes did not occur in control subjects.
Animals
In ketamine-treated rats chronically exposed to ACTH, plasma C-reactive protein (CRP), a general marked of inflammation, was nonsignificantly elevated compared to non-stressed rats and ACTH-exposed animals not given ketamine (Walker et al., 2015). Ketamine (10 mg/kg IP) reduced FST immobility at 1 h in control rats and in ACTH-exposed rats. Plasma CRP was higher in ACTH-treated animals compared to control animals, and ketamine-responsive ACTH animals had higher CRP that ACTH animals not given ketamine. There was a nonsignificant increase in plasma CRP among ketamine-responsive ACTH animals compared to nonresponders.
Nonresponsive rats had lower plasma TNF-α compared to ACTH animals not treated with ketamine and ketamine responders had nonsignificantly higher plasma TNF-α compared to nonresponders (Walker et al., 2015). Ketamine did not affect IL-6.
Depressive behavior and elevated pro-inflammatory cytokine levels in the hippocampus caused by CUMS were reversed by ketamine (10 mg/kg IP) in rats (Wang et al., 2015). Compared to control animals, CUMS-exposed rats had increased FST immobility and increased latency to feed in the NSFT; both effects were attenuated by ketamine. CUMS also increased hippocampal IL-1β, IL-6, and TNF-α. Ketamine reversed the increase in IL-1β at 30 min and 1 h; the increase in IL-6 at 30 min, 1 h, 2 h, and 4 h; and the increase in TNF-α at 2 and 4 h. CUMS also increased hippocampal IDO and the kynurenine/tryptophan ratio, effects that were reversed by ketamine at all timepoints.
R-ketamine (10 mg/kg IP) had more potent behavioral effects than S-ketamine in the mouse CSDS model and it attenuated an increase in plasma RANKL in CSDS-susceptible animals (Zhang et al., 2018). Sucrose preference positively correlated with OPG/RANKL ratio. Plasma RANKL was lower in R-ketamine animals versus controls and the OPG/RANKL ratio was higher; there was no effect on OPN or the OPN/RANKL ratio. Only R-ketamine reduced immobility in the TST at 4 h and in the FST at 24 h in susceptible mice. In the SPT, the R-ketamine group had elevated sucrose preference compared to controls, while S-ketamine produced a smaller effect. Neither enantiomer had significant behavioral effects in non-stressed mice, nor did they affect plasma levels of inflammatory bone markers.
Opposing Studies
Humans
Kiraly et al. (2017) found TRD patients (n=33) had higher baseline serum IL-6 compared to healthy controls (n=26), but although there were transient changes in immune- and inflammation-related molecules after ketamine, no change correlated with its antidepressant effects. Four hours after ketamine (0.5 mg/kg IV, 40-min), serum IL-6 and IL-1α were modestly reduced; neither was affected at 24 h. In contrast, IL-7 was increased, IL-8 was decreased, and PDGF-AA was decreased at 24 h, but not at 4 h. Ketamine also reduced G-CSF, IL-13, and IP-10 at 4 h, but not 24 h.
Aside from the baseline elevation in IL-6 among TRD patients, levels of the chemokine MCP-1 and multiple colony-stimulating and growth factors were elevated at baseline versus controls, including G-CSF, GM-CSF, and PDGF-BB (Kiraly et al., 2017). The only difference between responders and nonresponders was in fibroblast growth factor 2 (FGF-2), which was lower at baseline in responders; baseline FGF-2 correlated with MADRS score change at 24 h.
An exploratory analysis of data from three trials of ketamine for treatment-resistant MDD (n=49) and BD (n=31) did not find correlations between cytokine changes and antidepressant effects following a single dose of 0.5 mg/kg (IV, 40-min) (Park et al., 2017). MDD patients were unmedicated, but BD patients could continue a stable mood stabilizer regimen. At 230 min, serum IL-6 and sTNFR1 were increased, but there was no effect on IFN-γ, IL-10, IL-2, or IL-8. Although baseline sTNFR1 correlated with depression severity, changes in that marker did not correlate with ketamine’s effects, nor did changes in IL-6.
Compared to MDD patients, BD patients had higher IL-6 and TNF-α after ketamine, and their sTNFR1 level was lower from baseline through Day 3 (Park et al., 2017). Levels of those markers in BD patients did not correlate with baseline mood severity and BD patients did not have a higher baseline MADRS score than MDD patients, so it is unclear if these differences are functionally important.
Relevant Ketamine Research
Humans
Ketamine administered during coronary artery bypass surgery did not affect cytokine levels compared to placebo (Cho et al., 2009). Ketamine (0.5 mg/kg IV, 40-min) was given to 25 patients, while saline was administered to 25 control subjects. CRP and IL-6 increased to a similar extent in both groups, while TNF-α was unaffected. This suggests that IL-6 elevations observed in ketamine studies should not be immediately interpreted as relevant unless ketamine’s effect is different from placebo.
Animals
There was a reduction in brain (PFC and hippocampus) IL-1β and IL-6 alongside reduced FST immobility at 30 min in male rats treated with ketamine (10 mg/kg IP), but there was no investigation of a potential influence of cytokine change in the antidepressant-like effect (Yang et al., 2013).
Relevant Non-Ketamine Studies
In 16 patients with chronic hepatitis C who were psychiatrically healthy at baseline, treatment with the pro-inflammatory cytokine IFN-α had depressive effects (Wichers et al., 2005). MADRS score and the ratio of kynurenine/tryptophan, an indicator of IDO activity, increased during 24 weeks of treatment; the ratio of kynurenine to kynurenic acid (KA) also increased, potentially indicative of neurotoxicity since KA has anti-excitotoxic effects. 31% of patients met criteria for MDD at least once during the study; MADRS score correlated with kynurenine/KA ratio. The tryptophan/CAA (competing amino acid) ratio did not change, indicating tryptophan availability in the brain was not affected, and tryptophan/CAA did not correlate with depressive symptoms.
The increase in kynurenine/tryptophan ratio observed by Wichers et al. (2005) is neurologically and psychiatrically relevant because it may reflect a greater likelihood of neurotoxicity. The endogenous enzyme IDO converts the serotonin precursor tryptophan to kynurenine and kynurenine is further metabolized to an array of molecules, some of which are neurotoxic, like quinolinic acid and 3-hydroxykynurenine. The risk of IDO-mediated neurotoxicity may be countered by a different kynurenine metabolite, kynurenic acid, which is an NMDAR antagonist that may attenuate NMDAR activation caused by quinolinic acid.
Lindqvist et al. (2009) found IL-6 in the CSF of suicide attempters (n=63) was higher than in healthy controls (n=47) and those who engaged in violent suicide attempts had the highest IL-6. MADRS score positively corelated with IL-6 across all patients. In analyzing potential effects on the monoaminergic system, the authors found IL-6 and TNF-α positively correlated with 5-HIAA (serotonin metabolite) and HVA (dopamine metabolite), but not with MHPG (norepinephrine metabolite).
Plasma IL-6 was also elevated in a sample of 47 suicide attempters compared to 17 non-suicidal MDD patients and 16 healthy controls (Janelidze et al., 2011). Additionally, suicide attempters had elevated TNF-α and reduced IL-2 compared to the other groups, with the difference remaining significant after controlling for severity of depression, anxiety, and medication use. Non-suicidal MDD patients were unmedicated at the time of analysis, but 41/47 suicide attempters were on medications.
Quinolinic acid, but not kynurenic acid, was higher in the CSF of unmedicated suicide attempters (n=64) compared with control subjects (n=36) (Erhardt et al., 2013). The elevation in quinolinic acid correlated with increased CSF IL-6 and with total Suicide Intent Scale score, i.e. higher quinolinic acid correlated with worse suicidality. When the suicide attempters were re-assessed six months later, there was a significant decrease in quinolinic acid, suggesting it may be acutely associated with active suicidality.
7.8 VEGF
Vascular endothelial growth factor (VEGF) contributes to hippocampal neurogenesis in vitro and in animals via activation of its receptor VEGFR-2 (aka kinase insert domain receptor; KDR; Flk1), which may play a role in stress-related depressive behavior (Clark-Raymond and Halaris, 2013). It is more difficult to evaluate its relevance to depression in humans and the literature is currently inconclusive, with some studies showing reduced peripheral VEGF in depression and others showing an increase. Likewise, some studies report increased VEGF during antidepressant use, while others show no effect.
Pisoni et al. (2018) found reduced serum VEGF-C and BDNF in TRD patients (n=36) compared to control subjects (n=36), whereas angiopoietin-1 was increased. Lower VEGF-D also predicted treatment nonresponse and nonresponders had a reduction in VEGF and VEGF-C during treatment, whereas there was no change in responders.
In rodents, the rapid and sustained antidepressant effects of an intra-mPFC infusion of BDNF were blocked when a VEGF neutralizing antibody was also infused, and mPFC neuron-specific deletion of VEGF attenuated the effects of BDNF (Deyama et al., 2019). BDNF exposure increased VEGF release and enhanced dendritic complexity in vitro, effects that were attenuated by a VEGFR-2 antagonist. BDNF and VEGF seem to be involved in each other’s effects, as an intra-mPFC infusion of VEGF also produced antidepressant-like effects that were prevented by blocking BDNF.
Supporting Studies
A single dose of ketamine (10 mg/kg IP) increased the number of BrdU+ cells (primarily neurons) in the hippocampal DG region of rats at 24 h, indicating an increase in neurogenesis shortly after administration (Choi et al., 2016). This was accompanied by antidepressant effects in the FST and NSFT at 24 h, as well as increased hippocampal VEGF expression. The opposite effects were produced by hippocampal knockdown of VEGF, which caused depressive-like behavior and reduced hippocampal neurogenesis; ketamine partially attenuated both changes. VEGF knockdown also attenuated ketamine’s antidepressant effects and induction of hippocampal neurogenesis.
Consistent with those results, Deyama and colleagues (2019) found the antidepressant effects of ketamine (10 mg/kg IP) in rodents were blocked by deletion of VEGF or VEGFR-2 specifically in excitatory forebrain neurons or by the intra-mPFC infusion of a VEGF neutralizing antibody. When the antibody was infused 30 min before systemic ketamine, the behavioral effects of ketamine in the FST and NSFT were blocked 24 h and five days later. Ketamine increased neuronal activation, as shown by elevated c-Fos expression in rat mPFC, which was attenuated by VEGF neutralization.
Infusion of VEGF into the mPFC mimicked ketamine’s antidepressant effects and its efficacy was blocked by neuron-specific deletion of VEGFR-2 (Deyama et al., 2019). Both ketamine and VEGF had neurotrophic and synaptogenic effects. Ketamine (500 nM) and VEGF applied to rat primary cortical cultures increased dendrite branch crossings at 24 h, an effect blocked by the VEGFR-2 inhibitor ZM323881. In rats, ketamine (10 mg/kg IP) increased spine density in the apical tuft of infralimbic and prelimbic mPFC layer V pyramidal neurons at 24 h, which was blocked by VEGF deletion. This suggests VEGF and its signaling through VEGFR-2 are important for ketamine’s antidepressant, neurotrophic, and synaptogenic effects.
7.9 Opioid Receptors
Background
Opioids are known for their mood elevating, analgesic, and anxiolytic effects, but because they have a high abuse potential they have largely been kept out of modern psychiatry. Morphine was administered for depression, anxiety, and insomnia in the 1800s and early 1900s. However, criticism of the practice grew through the early part of the 20th century leading to the abandonment of classic opioids for those conditions (Weber and Emrich, 1988). Since the mid-1950s, opioids have mostly been restricted to use in surgery and pain with only a few exceptions, like the antidepressant tianeptine.
Faced with inadequate efficacy from standard medications, the psychiatric medicine field has developed a cautious interest in opioids in recent years (Lutz and Kieffer, 2013). For example, the MOR partial agonist buprenorphine is now used in some cases of TRD and in people experiencing opioid addiction concurrent with depression. Research has also implicated the endogenous opioid system in mood disorders. For example, endogenous opioids (i.e. enkephalins and endorphins) have antidepressant properties in animals, whereas the opioid receptor antagonist naloxone has depressive effects in the LH model. Consistent with those results, inhibitors of enkephalin breakdown (i.e. enkephalinase inhibitors) have antidepressant-like effects.
A literature review of buprenorphine for TRD found evidence supporting the use of low doses in that condition, although there is only a small body of evidence and the effects are not substantially superior to other adjunctive TRD treatments (Stanciu et al., 2017).
Ketamine seems to interact with opioid receptors, though few studies have explored potential connections between the opioid system and ketamine’s antidepressant effects. S-ketamine has stronger direct interactions with opioid receptors, yet R-ketamine is more potent as an antidepressant in animals. This suggests that if the opioid system is involved in ketamine’s mechanism, it may be through indirect modulation. Alternatively, the opioid system may not be involved or it may only have to be intact for the expression of ketamine’s benefits.
Supporting Research
Humans
A paper published in 2018 by Williams and colleagues generated a lot of interest and skepticism regarding the role of opioid receptors. Their DBRCT crossover trial of 30 TRD patients, which was stopped after obtaining data from 14 people, studied ketamine (0.5 mg/kg IV, 40-min) combined with either placebo or naltrexone (50 mg oral) pretreatment (Williams et al., 2018). Patients could continue treatment with antidepressants that were considered unlikely to interact with ketamine. One day after treatment, the ketamine-only group had a 58% (7/12) response rate; those improvements were reduced, though not eliminated, by naltrexone on Days 1 and 3. Naltrexone did not affect ketamine-induced dissociation Differences between the groups were seen up to Day 3, but they did not persist to Day 5 and beyond. On Day 1, remission was present in 5/7 responders with ketamine-only treatment, but 0/7 had remission when naltrexone was also given. Ketamine still had an effect on Day 1 in the presence on naltrexone when assessed with the HDRS-17, but no significant effect was observed with the 6-item HDRS or the MADRS, which are more reflective of core depressive symptoms. Therefore, other measures of depression found that naltrexone eliminated ketamine’s efficacy.
There were a number of responses to this paper. Dr. Mark George, Professor of Psychiatry at the Medical University of South Carolina, encouraged a cautious interpretation of the results, arguing that the study did not conclusively show that ketamine is an antidepressant merely because it is an opioid (George, 2018). Echoing that critique, Dr. Gerard Sanacora, Professor of Psychiatry at Yale University, noted the results may just mean normal functioning of the endogenous opioid system is necessary for the antidepressant effects. Further, although studies have not shown clear depressogenic effects of naltrexone itself, that does not prohibit it from generally impeding antidepressant activity from other drugs (Sanacora, 2019).
One of the concerns repeatedly alluded to or explicitly mentioned in the responses to Williams et al. (2018) is that labeling ketamine an opioid could be consequential at a time when there are major concerns about opioid misuse and addiction. In their original paper, the authors argued the results provide “strong justification for further caution against widespread and repeated use of ketamine before further mechanistic testing has been performed” (Williams et al., 2018).
In a response to Sanacora (2019), the authors conceded that their results can be interpreted in multiple ways, but they also doubled down on concerns about ketamine’s safety (Heifets et al., 2019). This included venturing into poorly supported territory by claiming “the epidemiological evidence speaks for itself: ketamine can be highly reinforcing, lends itself to abuse, and may result in significant adverse medical and behavioral sequelae.” The position expressed by Heifets et al. (2019) is a somewhat alarmist interpretation of the literature. Ketamine use can be compulsive and/or dangerous in a minority of users, but the risks are likely small in medical settings considering its already modest abuse potential in nonmedical settings.
Opposing Research
Humans
In an 8-week open-label trial, ketamine was provided to five MDD patients with comorbid alcohol use disorder (Yoon et al., 2019). They were alcohol-abstinent for at least five days before the initial ketamine dose. Injectable naltrexone (380 mg) was given 2-6 days before the start of ketamine (0.5 mg/kg IV, 40-min), which was administered once per week for four weeks. Depressive symptoms were reduced at 40 min, 240 min, and Day 1, but not Day 7.
The ketamine + naltrexone condition produced a response in 3/5 after the first dose and in 5/5 by the last dose, although one patient left the trial after only two infusions (Yoon et al., 2019). The importance of the response in 3/5 patients after the first infusion is hard to determine since 3/5 patients also experienced at least some depression alleviation between baseline and shortly before the first infusion. Depressive symptoms were reduced by 57-92% after the last infusion and 4/5 patients experienced reduced alcohol craving and consumption.
Animals
The opioid receptor antagonist naltrexone failed to block ketamine’s antidepressant effects in the CSDS and LPS models in mice (Zhang and Hashimoto, 2018). Ketamine (10 mg/kg IP) attenuated a CSDS-induced increase in FST immobility (at 24 h) and TST immobility (same day) in susceptible mice, and it increased sucrose preference at 48 h. Naltrexone did not block those effects or produce antidepressant-like activity on its own. It also failed to attenuate the antidepressant effects of ketamine in the LPS model.
Relevant Ketamine Research
Humans
In a DBRCT of naltrexone (25 mg oral) preadministration, some ketamine-induced subjective effects were changed, but it did not cause widespread antagonism (Krystal et al., 2006). One group of healthy people (n=31) received a perceptual dose of ketamine (0.23 mg/kg IV bolus, then 0.58 mg/kg over 1 h) or placebo and the other (n=24) received a subperceptual dose (0.081 mg/kg IV 10-min; then 0.4 mg/kg over 1 h). Ketamine dose-dependently increased PANSS ratings for schizophrenia-related positive symptoms, negative symptoms, emotional discomfort, and cognitive effects. The smaller dose had a subjective effect that was likened to two drinks of alcohol, while the higher dose produced an experience more akin to five drinks. Naltrexone alone did not have effects; however, it potentiated the increase in PANSS score caused by the lower ketamine dose, but not the higher dose.
Animals
Smith et al. (1985) reported that the antinociceptive effect of ketamine (160 mg/kg IP), a dosage that increased latency in the tail-flick assay, was antagonized by naloxone in rats. The antinociceptive effect was blocked by systemic naloxone, but naloxone administered directly to the periaqueductal gray (PAG) region of the CNS was ineffective despite the authors hypothesizing ketamine may activate pain inhibition pathways that extend down the spinal cord from the PAG. In contrast, intra-PAG naloxone did block the effect of morphine.
Analgesic doses of ketamine (80-120 mg/kg IP) interacted with opioid receptors in rats, as shown by reduced naloxone binding in the brain and spinal cord (Smith et al., 1987). With the exception of the cerebellum, all brain regions were affected, with significant effects in the cortex, hippocampus, thalamus, and striatum. Ketamine was maximally active at 120 mg/kg, which reduced naloxone binding by 68% in the brain and 55% in the spinal cord. It had no effect at 40 mg/kg, a dosage that is more relevant to its antidepressant properties.
Two weeks after chronic ketamine treatment (30 mg/kg IP for 10 days), male rats had an increased pain threshold in a tail root stimulation test if they were housed alone, but group-housed animals did not show an effect (Becker et al., 2006). Likewise, the antinociceptive effect of morphine was altered by social isolation. Group-housed and singly-housed rats both had reduced MOR binding in the hippocampus; in contrast, MOR binding was increased in the frontal cortex exclusively in singly-housed rats.
The cognitive effects of ketamine (20 mg/kg IP) were similar to those of the KOR agonist salvinorin A in male rats and its activity was partially attenuated by a KOR antagonist (Nemeth et al., 2010). At 20 mg/kg, ketamine impaired cognitive performance by decreasing the likelihood of rats providing a response in a reaction time task and it increased the latency for correct responding; there was a trend-level effect at 10 mg/kg. Like ketamine, salvinorin A impaired task performance by increasing omissions, not reducing accuracy.
All salvinorin A effects were blocked by the KOR antagonist JDTic, while for ketamine, it blocked the effect on correct responding and attenuated the increase omissions, but it failed to attenuate the increase in latency for correct responding (Nemeth et al., 2010). In vitro experiments showed ketamine was a relatively weak ligand for human KOR (Ki = 25 μM) and it had an EC50 of 29 μM. By comparison, its affinity for rat σ receptors was 5.2 nM and its rat NMDAR affinity was 890 nM. Ketamine functioned as a full agonist at KOR; its effects were blocked by JDTic.
Social isolation reduced the affinity of the MOR agonist DAMGO in the cerebral cortex of saline-treated rats, an effect attenuated by 14 days of ketamine (30 mg/kg/d IP) (Kekesi et al., 2011). In hippocampal membranes, MOR Bmax (receptor concentration) was increased by ketamine, which was attenuated by social isolation; isolation and ketamine did not affect binding affinity in that region. Ketamine, but not isolation, increased binding capacity in the spinal cord in non-isolated animals. Social isolation reduced MOR efficacy (Emax) in the spinal cord, whereas ketamine increased it; this was assessed by measuring DAMGO-stimulated GTPγS binding, which is indicative of receptor activation.
Ketamine (4 μg ICV) caused antinociception in mice that was antagonized by naloxone, the MOR antagonist clocinnamox, and the DOR antagonist naltrindole, but not by the KOR antagonist nor-binaltorphimine (Pacheco et al., 2014). To test the influence of endogenous opioids, the aminopeptidase inhibitor bestatin was administered to prevent enkephalin from being cleaved at its tyrosyl-glycine bond. Bestatin enhanced the antinociceptive effect of low-dose ketamine, which was attenuated by naltrindole and clocinnamox. This implicates MOR- and DOR-mediated activity in the antinociception produced by ketamine; further, endogenous opioids may be involved.
In Vitro
In an assay using rat brain homogenate and the radioligand dihydromorphine, ketamine was shown to be a ligand for opioid receptors (Finck and Ngai, 1982). It had an IC50 of 23 μM; S-ketamine had a higher affinity (IC50 = 16 μM) than R-ketamine (IC50 = 46 μM). Similar to known opioids, ketamine inhibited contractions in the guinea pig ileum assay. S-ketamine was twice as potent as R-ketamine, although naloxone only partially antagonized its effect. Ketamine’s IC50 values were lower in the pig ileum assay: 4.89 μM for the racemate, 3.42 μM for S-ketamine, and 6.7 μM for R-ketamine. It also displaced the opioid etorphine in mouse brain, specifically in the thalamic region, whereas it was ineffective in the cortex.
Dihydromorphine’s highest affinity site is MOR, but it also has an appreciable affinity for DOR and KOR (Finck and Ngai, 1982). The relatively nonselective nature of dihydromorphine could have obscured ketamine’s potency since ketamine may be selective for only one or two opioid receptors. Antidepressant concentrations of ketamine would have minimal effect on opioid receptors if the IC50 values from the radioligand binding assay are applicable, but opioid receptor activity could be more relevant if the values from the pig ileum assay are more in line with its affinity in humans.
Contradicting the results reported by Finck and Ngai (1982), ketamine appeared to have very weak interactions with opioid receptors based on the guinea pig ileum assay, where its IC50 was 2100 μM (Bansinath et al., 1992). The effect of ketamine was partially blocked by naloxone and by the KOR antagonist nor-binaltorphimine.
Interactions between ketamine and MOR were reported in SK-N-SH (human neuroblastoma cell line expressing MOR) and CHO cells expressing recombinant MOR (Gupta et al., 2011). CHO cells treated with ketamine (10 μM) had a 3.5-fold increase in ERK1/2 phosphorylation, while morphine caused a 9.6-fold increase and fentanyl produced a 13.2-fold increase. Combining ketamine with morphine or fentanyl greatly increased their effect, with an 18.8-fold increase for the morphine combo and a 28.6-fold increase for the fentanyl combo; morphine and fentanyl did not synergize.
Ketamine also dose-dependently increased p-ERK1/2 in SK-N-SH cells, with an EC50 of 7 μM, much weaker than morphine (EC50 = 5.8 nM) or fentanyl (EC50 = 63 nM) (Gupta et al., 2011). In terms of Emax (i.e. max elevation in p-ERK1/2), ketamine increased phosphorylation 2.7-fold, morphine had a 4.4-fold increase, and fentanyl had an 8.1-fold increase. Synergism was observed when ketamine was added to either opioid. The importance of MOR was tested using the selective antagonist CTOP, which blocked ketamine-induced potentiation as well as morphine and fentanyl-induced ERK1/2 phosphorylation.
The addition of ketamine increased the effect of morphine (1 nM) from 2.6-fold to 28.7-fold, while the effect of fentanyl (1 nM) was increased from 1.8-fold to 33.3-fold (Gupta et al., 2011). A substantial interaction was still seen with a smaller concentration of ketamine (10 nM), which increased the effect of morphine (100 nM) from 3.2-fold to 8.9-fold. That interaction did not involve altered MOR binding, as ketamine (10 μM) did not affect morphine binding. Potential effects upstream of ERK1/2 were evaluated by examining opioid-induced GTPγS binding in SK-N-SH cells. Ketamine increased the EC50 (i.e reduced the potency) of morphine from 25 to 299 nM and of fentanyl from 6.7 to 61 nM; it also caused a trend-level reduction in the maximum opioid-mediated increase in GTPγS binding. Because ketamine had an opposite effect upstream of ERK1/2, the interaction point is likely elsewhere.
Potentially relevant to its role in pain treatment, ketamine impeded the desensitization of ERK1/2 signaling after opioid exposure (Gupta et al., 2011). On their own, morphine and fentanyl increased ERK1/2 phosphorylation for 30 min, but the increase persisted for >90 min when ketamine was added. Ketamine also sped up resensitization from 30 min to 15 min, as determined by inducing desensitization with 30-min of opioid treatment and then assessing how long it took for opioids to again increase p-ERK1/2.
Relevant Non-Ketamine Research
Humans
ALKS 5461, a combination of sublingual buprenorphine with the MOR antagonist samidorphan, had some signs of efficacy in TRD patients (n=142) (Fava et al., 2016). Two active treatment groups were compared with placebo: one group received 2 mg of each drug (the 2/2 group) and the other received 8 mg of each (8/8 group). During the 4-week trial, symptom improvement was superior in the 2/2 group, based on MADRS, HDRS, and CGI-S scores; the 8/8 group only had trend-level effects. The HDRS response rates at the end of treatment were: 26% with placebo, 47% with 2/2, and 36% with 8/8.
Severely suicidal patients who received buprenorphine had greater symptom alleviation than patients treated with placebo (Yovell et al., 2016). In a DBRCT manner, ultra-low-dose sublingual buprenorphine (n=40) was compared with placebo (n=22); concurrent medication use was permitted. Buprenorphine was initiated at 0.1 mg 1-2x daily, increasing to an average final dose of 0.44 mg/d. Buprenorphine patients had larger reductions in suicidality (BSS score) at Weeks 2 and 4; the effects were not altered by concurrent medication use or BPD diagnosis. Withdrawal was not reported after discontinuing therapy.
Animals
Tianeptine, which has been an approved treatment for depression in some countries for years, was found to produce acute and chronic antidepressant-like effects in mice that were dependent on MOR agonism (Samuels et al., 2017). Its primarily metabolite has similar MOR-dependent effects on depressive behavior. Unlike morphine, it did not show signs of tolerance or withdrawal.
7.10 Gut Microbiome
The gut microbiome includes 1800 phyla and 40,000 bacterial species, with the most prominent phyla including Firmicutes (e.g. Lactobacillus) and the Bacteroidetes (e.g. Bacteroides); among the other major phyla are Proteobacteria, Actinobacteria, and Cyanobacteria (Sherwin et al., 2016). Some gut microbes appear to influence metabolism, CNS disorders, and the immune system.
There is some correlative evidence that suggests ketamine’s antidepressant effects could partly involve changes to the gut microbiome and the gut microbiota-brain axis. This evidence builds upon research implicating gut microbiota in mood and other aspects of cognitive and psychological functioning, but a role for gut microbiota in ketamine’s mechanism still needs more support.
Experiments in animals have shown that eliminating gut microbes can produce antidepressant and anxiolytic-like behavior, and human studies have demonstrated gut microbiota can be immunomodulatory, potentially impacting inflammatory cytokine activity in the brain. This is particularly relevant when the intestinal epithelial barrier is weakened, which can facilitate the movement of microbes from the intestines to the body (Macedo et al., 2017; Zheng et al., 2016). Ketamine and other antidepressants, including some MAOIs, TCAs, and SSRIs appear to have antimicrobial effects; meanwhile, antimicrobials can produce neurological changes, such as increased expression of the glutamate transporter GLT-1 (EAAT2) (Macedo et al., 2017).
Supporting Studies
Animals
Chronic stress altered the gut microbiome of male mice and those changes were partially reversed by ketamine (10 mg/kg IP), with R-ketamine showing greater potency than S-ketamine (Yang et al., 2017). Fecal samples were taken four days after drug treatment. In CSDS-susceptible mice, both ketamine enantiomers reduced immobility and increased sucrose preference, though R-ketamine was more potent. Analysis of gut microbiota at the phylum level showed a reduction in Tenericutes and increase in Actinobacteria in CSDS-susceptible mice; ketamine did not attenuate either change.
However, at the class level, susceptible mice had a reduction in Deltaproteobacteria that was attenuated by both enantiomers (Yang et al., 2017). Mollicutes were decreased in susceptible mice, which was only reversed by R-ketamine. At the family level, Desulfovibrionaceae were increased in susceptible mice; S-ketamine enhanced the increase, whereas R-ketamine had no effect. And at the genus level, Butyricimonas were reduced in susceptible mice, which both enantiomers attenuated, though with greater potency from R-ketamine.
If the changes observed by Yang and colleagues (2017) are relevant to the antidepressant effects of ketamine, R-ketamine would be expected to exhibit greater potency, and changes in Mollicutes and Butyricimonas could be particularly relevant since they were differentially affected by the enantiomers.
Compared to the NMDAR antagonist lanicemine, R-ketamine (10 mg/kg IP) more effectively reversed depressive behavior in CSDS-susceptible mice and it had distinct effects on the gut microbiome (Qu et al., 2017). R-ketamine, but not lanicemine, was effective in the TST, FST, and SPT. Similarly, only R-ketamine attenuated changes in the levels of Bacteroidales, Clostridiales, and Ruminococcaceae in susceptible mice. At the genus level, R-ketamine was more potent for attenuating an increase in Clostridium caused by CSDS exposure.
FST immobility after LPS exposure was reduced by ketamine (10 mg/kg IP) in mice (Huang et al., 2019). LPS significantly altered the gut microbiome and levels of some microbes correlated with ketamine’s effect. The phylum Actinobacteria and the class Coriobacteriia correlated with FST immobility, suggesting they may be biomarkers for ketamine-induced antidepressant effects in inflammatory models.
Relevant Ketamine Research
Animals
Exposure to ketamine (2.5 mg/kg/d IP) for one week changed the gut microbiome of male rats (Getachew et al., 2018). Analysis one day after the end of treatment showed an increase in Lactobacillus, Turicibacter, and Sarcina, whereas Mucispirillum and Ruminococcus were reduced by ketamine.
Relevant Non-Ketamine Research
Humans
Analysis of fecal samples from active MDD patients (n=29) compared with patients who had responded to treatment (n=17) and healthy controls (n=30) found the active MDD patients had increased fecal bacterial α-diversity (i.e. diversity of species) compared to control subjects, but not compared to responded-MDD patients (Jiang et al., 2015). Compared to controls, active-MDD patients had increased levels of the phyla Bacteroidetes, Proteobacteria, and Actinobacteria, whereas Firmicutes were lower. Although individual patients differed substantially from each other, group differences were detected, including higher levels of Enterobacteriaceae and Alistipes, and lower Faecalibacterium compared to controls. Patients with more severe depressive symptoms had lower levels of Faecalibacterium.
Gut bacteria can affect inflammation, but inflammatory markers (TNF-α, IL-6) did not differ between patients and controls; however, BDNF was reduced among patients (Jiang et al., 2015). Clostridium bacteria in the XIVb subcluster negatively correlated with serum BDNF.
The absence of gut microbes in mice was associated with lower FST immobility and when fecal microbiota from MDD patients (n=5) was transplanted to the microbe-free mice, depressive behavior was elevated two weeks later compared with mice that received fecal microbiota from healthy subjects (n=5) (Zheng et al., 2016). MDD patients had different levels of Firmicutes, Actinobacteria, and Bacteroidetes compared to controls. Mice that received transplants from depressed humans exhibited changes in microbial genes and host metabolites related to carbohydrate and amino acid metabolism, suggesting they may influence behavior through altered metabolism.
Animals
In the mouse CSDS model, administration of an anti-IL-6 antibody (MR16-1) produced rapid and persistent antidepressant effects in CSDS-susceptible mice; intravenous injection was effective, but ICV injection was not (Zhang et al., 2017). The antibody normalized dendritic spine density in susceptible mice as well as synaptic protein expression (i.e. PSD95 and GluR1). It also attenuated a reduction in Oscillospira and Firmicutes/Bacteroidetes ratio seen in susceptible mice. Given these effects, altered peripheral IL-6 may have affected the gut microbiome, potentially influencing mood.
7.11 Sex Differences
Sex has not been found to influence the efficacy of ketamine in patients with mood disorders, but preclinical evidence suggests there may be some behavioral and mechanistic differences between males and females. As with much of preclinical research, animal studies on ketamine have primarily used males, yet the sexes do not seem to have identical responses to the drug. Female rodents may be more sensitive to ketamine than males, although their response may also be more transient (Carrier and Kabbaj, 2013; Franceschelli et al., 2015).
Supporting Studies
Animals
Female rats were more sensitive than males in some behavioral tests, which was associated with estrogen and progesterone activity, but not with mTOR or eEF2 (Carrier and Kabbaj, 2013). Ketamine (2.5, 5, 10 mg/kg IP) given 30 min before the FST reduced immobility at all doses in females, but male rats only responded to 5 and 10 mg/kg. Conversely, only male rats responded in the SPT at 48 h, where 5 and 10 mg/kg increased sucrose preference. Because a depressive state was not initially induced to alter behavior in these tests, it is unclear how to interpret the differences. Neither sex responded in the EPM, which measures anxiety-like behavior.
Ovariectomy (removal of the ovaries) increased basal FST immobility in female rats and reduced ketamine’s effect, with 2.5 mg/kg no longer being active in the FST (Carier and Kabbaj, 2013). Administration of either estrogen or progesterone failed to restore normal ketamine response in ovariectomized females, but a combination of the two was effective. Interestingly, male rats had a reduction in p-eEF2 in the hippocampus with 5 mg/kg, but that dose did not affect p-eEF2 in females. mPFC synaptoneurosomes showed an increase in p-mTOR with 5 mg/kg in both sexes.
The behavioral and neurochemical effects of ketamine were greater in female mice at 30 min and 24 h, but a more persistent effect was observed in males (Franceschelli et al., 2015). Females responded to as little as 3 mg/kg (IP) in the FST at 30 min, whereas males required ≥5 mg/kg; at 24 h, males only responded to 10 mg/kg, but females responded to 5 and 10 mg/kg. Ketamine (10 mg/kg) rapidly reduced hippocampal glutamate in male mice and increased aspartate in the PFC of females; the 5-HIAA/serotonin ratio at 24 h was reduced in females, but there was only a nonsignificant reduction in males.
Both sexes exhibited anhedonic behavior after CMS exposure and ketamine was ineffective for alleviating those changes, as shown in the SPT on Day 6; CMS-induced anxiety-like behavior in the marble burying test was unaffected by ketamine (Franceschelli et al., 2015). Persistent antidepressant effects in the FST (Day 7) and splash test (Day 5) only occurred in male CMS-exposed mice.
Thelen et al. (2016) studied the effects of a frequent dosing regimen that is mostly irrelevant to acute antidepressant effects, though it demonstrated differences between the sexes; mice received daily ketamine (3, 5, 10 mg/kg IP) for 21 days. Chronic ketamine caused antidepressant-like effects in males when they were tested 24 h after the last injection, but females exhibited depressive behavior and a more anxious phenotype (e.g. reduced time in the center of the OFT arena). The antidepressant effects in males correlated with increased hippocampal synaptic protein expression (synapsin I and SNARE) and serotonin turnover, while females had reduced glutamate and aspartate levels. Synapsin I positively correlated with hippocampal glutamate in males, but not females.
7.12 Regions of Interest
Lateral habenula (LHb)
The lateral habenula (LHb) regulates reward pathway activity by targeting dopaminergic neurons in the VTA and is itself targeted by stress-relevant limbic and cortical regions. LHb neurons are activated by aversive stimuli and overactivity of the LHb may be associated with depression.
Supporting Research
Animals
Ketamine (25 mg/kg IP) administered to congenitally learned helpless (cLH) rats reduced FST immobility at 1 h, which was mediated by inhibition of NMDAR-dependent bursting activity in the LHb (Yang et al., 2018). LHb neurons in depressed animals exhibited more bursting activity and theta-band synchronization, changes that were attenuated by ketamine; burst activity in the LHb may be involved in despair and anhedonia. Bursting activity depended on NMDARs and low-voltage-dependent T-type calcium channels (T-VDCCs) since locally blocking either in the LHb was sufficient to cause rapid antidepressant-like effects.
The T-VDCC blocker ethosuximide had antidepressant-like effects (FST and SPT) in chronic restraint stress mice (Yang et al., 2018). Likewise, local application of the nonselective calcium channel blocker mibefradil in the LHb of cLH rats produced antidepressant-like effects.
Relevant Non-Ketamine Research
Humans
A patient with severe TRD experienced sustained remission beginning four months after the initiation of deep brain stimulation of the LHb; the delay may indicate the involvement of synaptic plasticity (Sartorius et al., 2010).
Animals
Excitatory synapses onto LHb neurons projecting to the VTA are potentiated in rat LH models of depression (both acute LH and congenital LH) (Li et al., 2011). Synaptic potentiation along that pathway was associated with increased presynaptic release and it correlated with helplessness behavior. In contrast, reducing synaptic activity onto LHb neurons that project to the VTA by depleting neurotransmitters attenuated depressive behavior.
Nucleus accumbens (NAc)
Relevant Non-Ketamine Research
Animals
Chronic social defeat stress (CSDS) increased c-Fos induction (an indicator of neuronal activity) in VTA dopamine neurons and in their target NAc neurons, and CSDS-susceptible mice showed sensitization to c-Fos induction when they were re-exposed to a social target four weeks after the CSDS procedure (Berton et al., 2006). Stress was associated with increased BDNF in the NAc one day and four weeks later; BDNF gene deletion in VTA neurons had an antidepressant-like effect, suggesting the increase in NAc BDNF caused by stress comes from VTA dopaminergic neurons projecting to that region. Acute fluoxetine and imipramine did not improve CSDS-induced social interaction deficits, but chronic treatment did. The benzodiazepine chlordiazepoxide was ineffective with acute and chronic exposure, suggesting the behavior change involves despair and defeat, not anxiety.
Hippocampus
Supporting Research
Animals
Activity within the ventral hippocampus to mPFC (vHipp-mPFC) pathway was important for ketamine-induced antidepressant effects in male rats and increased plasticity along that pathway involved BDNF-TrkB activity (Carreno et al., 2015). Ketamine (10 mg/kg IP) reduced FST immobility at 30 min and 1 week; the 1 week effect was lost when the vHipp was transiently inactivated using lidocaine concurrently with ketamine injection. TrkB phosphorylation increased 30 min after ketamine, but the effect was absent by one week. Bilateral administration of the TrkB inhibitor K252a to the vHipp prior to ketamine injection blocked ketamine-induced TrkB phosphorylation and behavioral effects.
Optogenetic stimulation of the vHipp-mPFC also had antidepressant-like effects when the GABAA receptor antagonist bicuculline was injected into the DRN to prevent hypothetical feedback from the mPFC to the DRN that would interfere with the FST results (Carreno et al., 2015). Furthermore, ketamine-like effects were produced when the mPFC neuron population that receives inputs from the vHipp was modified and activated by DREADD methodology. In contrast, DREADD-mediated activation of the NAc was ineffective, indicating vHipp-mPFC activity is uniquely involved in the alleviation of depressive behavior.
In male Flinders sensitive line (FSL) rats, a genetic model of depression, S-ketamine (15 mg/kg IP) reduced FST immobility at 24 h and increased hippocampal astrocyte size, number, and the length of astrocytic processes (Ardalan et al., 2017). At baseline, the volume of GFAP+ astrocytes in the CA1 stratum radiatum (SR) was smaller in FSL rats compared to Flinders resistant line (FRL) rats. Ketamine increased astrocyte size in the DG and nonsignificantly increased astrocyte volume in the CA1-SR of FSL rats.
Astrocytic branch length was increased one day after ketamine in FSL and FRL rats; greater astrocytic processes length and branch number correlated with reduced FST immobility (Ardalan et al., 2017). Ketamine increased the volume of the CA1 and granule cell layer of the DG one day after injection, and the increase in astrocyte size in the CA1-SR correlated with increased region size.
Opposing Research
Animals
Ketamine (10 or 50 mg/kg IP) and MK-801 reduced FST immobility in male rats at 30 min; the reduction was somewhat larger at 50 mg/kg (Engin et al., 2009). Anxiolytic effects were also observed in the EPM when rats were given diazepam, MK-801, or ketamine (50 mg/kg). A significant reduction in evoked theta activity in the hippocampus occurred after diazepam, fluoxetine, and MK-801 treatment, but ketamine (50 mg/kg) only had a trend-level effect and it did not affect theta power, whereas diazepam and MK-801 reduced it; activity was evoked by stimulating the reticular formation.
7.13 HCN Channels
Introduction
Hyperpolarization-activated, cyclic nucleotide-gated channel 1 (HCN1) knockdown in the hippocampal CA1 region produces antidepressant and anxiolytic-like effects in rats along with increased cellular excitability, BDNF expression, and p-mTOR in the dorsal hippocampus (Kim et al., 2012). Kim et al. (2018) reported an increase in HCN1 expression in rat dorsal CA1 neurons following CUS exposure; HCN1 knockdown blocked the depressive effects of CUS.
CSDS-susceptible mice displayed a significant increase in the firing rate of VTA dopaminergic neurons, which was attenuated by the HCN channel inhibitor cilobradine (Zhu et al., 2018). A single injection of cilobradine normalized social avoidance behavior in CSDS-susceptible mice acutely and persistently, with efficacy still observed 13 days later. The behavioral changes were associated with a reduction in pathological VTA dopaminergic neuron hyperactivity across the 13-day period.
Ketamine inhibits HCN1 in vitro at anesthesia-relevant concentrations, but subanesthetic doses may not have a large effect on channel activity. The EC50 for the racemate is 16 μM; S-ketamine is a stronger inhibitor of HCN1, with an EC50 of 7 μM (Chen et al., 2009). There was a large reduction in ketamine’s hypnotic effects in HCN1 KO mice.
Supporting Research
Animals
Ketamine-induced antidepressant effects in mice were lost when presynaptic HCN1 channels were inhibited or deleted, which was interpreted as occlusion of ketamine’s effects since HCN1 inhibitors on their own mimicked ketamine (Zhang et al., 2016). Inhibition of postsynaptic HCN1 channels in CA1 cells using ZD7288 did not prevent ketamine-induced enhancement of SC-CA1 EPSCs, supporting a role for presynaptic HCN1 channels instead. Postsynaptic HCN1 inhibition on its own did not increase the peak amplitude of SC-CA1 EPSCs even though it did reduce the Ih current mediated by HCN channels, along with reducing the decay time of EPSCs.
7.14 microRNA
microRNA (miRNA) are non-conding RNA (~22 nucleotides long) that regulate mRNA translation by binding to the untranslated 3’ region in a sequence-specific manner.
Supporting Research
Animals
Ketamine, fluoxetine, and ECT altered hippocampal miRNA expression in non-stressed male rats and had more effects in rats that were stressed; stress itself also affected miRNA expression (O’Connor et al., 2013). Ketamine was given acutely (10 mg/kg IP), while ECT was repeatedly administered for 10 days and fluoxetine was given for 21 days. Each treatment reversed some stress-induced changes and some targets overlapped, particularly with ketamine and ECT, which shared 43 miRNA targets. Microarray analysis identified a shared target in non-stressed rats (miR-598-5p) and a different shared target in stressed animals (miR-451); treatment normalized the stress-associated change in miR-451 expression.
Stress itself, specifically maternal separation (MS) early in life, affected 24 hippocampal miRNAs (O’Connor et al., 2013). Chronic fluoxetine decreased 4 miRNA (3 were a partial normalization of stress-induced changes), ECT changed 86 (48 decreased and 38 increased; 16 were a reversal of stress), and ketamine affected 55 (32 decreased and 23 increased; 11 were a reversal of stress). Since miR-598-5p and miR-451 were shared targets, those were analyzed further using qRT-PCR. miR-451 was reduced in stressed animals, but only fluoxetine reversed that change, contradicting the microarray findings. Fluoxetine and ECT increased expression of miR-598-5p in non-stressed animals, but ketamine was ineffective. This study therefore supports or goes against the role of miRNAs in the mechanism of ketamine depending on whether the microarray or qRT-PCR findings are correct. Interestingly, the pattern of changes caused by antidepressants in non-stressed animals was reminiscent of the pattern caused by stress, albeit with a smaller effect overall; this suggests antidepressants in healthy organisms may cause stress-like effects.
Hippocampal miRNAs in male rats were affected by ketamine (15 mg/kg IP), including one that regulates the BDNF gene, miR-206 (Yang et al., 2014). Ketamine reduced expression of 18 miRNAs and increased 22. miR-206 was downregulated in the hippocampus and in cultured neurons; overexpression of miR-206 attenuated ketamine-induced BDNF upregulation along with causing apoptosis in cultured neurons, which was reduced by ketamine (50 μM). In vitro, high expression increased calcium influx (HVA current) and reduced transient outward potassium currents in hippocampal pyramidal neurons; ketamine (50 μM) attenuated the calcium current.
An antidepressant dose of ketamine (10 mg/kg IP) in mice upregulated a cluster of hippocampal miRNAs associated with the 5-HT2C receptor: 448-3p, 764-5p, 1264-3p, 1298-5p, 1912-3p (Grieco et al., 2017). Those miRNAs are intronic and are located at the locus of the 5-HT2C receptor gene. The increase was abolished in GSK-3 knockin mice with constitutively active GSK-3, whereas the GSK-3 inhibitor L803-mts had antidepressant effects (LH and NSFT tests) and also upregulated the 5-HT2C receptor miRNA cluster in the hippocampus. Mice that were resilient to LH-induced depressive behavior had increased hippocampal miR-448-3p and miR-1264-3p, but depressed mice did not. Ketamine’s antidepressant effect was reduced by antagonism of miRNA-448-3p.
Expression of miR-29b-3p in the PFC was increased by ketamine (10 mg/kg IP) in rats, with no change in the hippocampus or hypothalamus; the increase was observed 1 to 12 h after ketamine (Wan et al., 2018). miR-29b-3p targets the metabotropic glutamate receptor 4 gene (GRM4). Rats with depressive behavior had a downregulation of miR-29b-3p and an upregulation of GRM4 in the PFC, while ketamine produced the opposite, i.e. increased miR-29b-3p expression and reduced GRM4 expression in the PFC of depressive rats and in primary neurons. Overexpression of miR-29b-3p enhanced cell survival, increased dendritic growth, increased extracellular glutamate, and inhibited apoptosis. In CUMS-exposed rats, overexpression reduced depressive behavior and GRM4 expression.
7.15 HDAC
Supporting Research
Animals
Ketamine increased hippocampal histone deacetylase 5 (HDAC5) phosphorylation (Ser259/Ser498) in male rats and in vitro (rat primary hippocampal neurons), yielding altered gene expression; the HDAC5 effect was important for its antidepressant-like effects (Choi et al., 2015). HDAC5 phosphorylation (at Ser259 and Ser498) was maximally increased at ~100 nM ketamine, with higher doses failing to show an effect; the impact of 100 nM peaked at 3-6 h and returned to baseline after 24 h. CaMKII and PKD, which are known to regulate HDAC5 phosphorylation, influenced ketamine’s effect. The CAMKII inhibitor KN-62 and the PKD inhibitor Go6976 attenuated the increase in HDAC5 phosphorylation.
Because phosphorylated HDAC5 is exported out of the nucleus, ketamine increased cytoplasmic p-HDAC5 (Choi et al., 2015). Nuclear export was dependent on ketamine-induced phosphorylation, as shown in hippocampal neurons containing HDAC5 with a serine-to-alanine mutation, which prevented export. In normal neurons, cytoplasmic translocation began by 30 min and subsided within 48 h. Consistent with research showing increased histone acetylation associated with HDAC5 nuclear export, ketamine increased acetylation of the histones H3 and H4.
Additionally, MEF2 transcriptional activity was increased by ketamine, an effect attenuated by inhibition of CaMKII or PKD; inhibitors of those kinases also blocked ketamine-induced upregulation of the MEF2 target genes Arc and Nurr77 (Choi et al., 2015). HDAC5 overexpression reduced MEF2 activity, which was reversed by ketamine. mRNA levels of the MEF2 downstream effector Klf6 were increased by ketamine, which was also sensitive to CaMKII and PKD inhibition. Like with p-HDAC5 nuclear export, a serine-to-alanine mutation in HDAC5 blocked ketamine’s MEF2 activation.
In rats, ketamine (10 mg/kg IP) increased hippocampal HDAC5 phosphorylation, with a peak effect at 6 h and the increase persisted at 12-24 h (Choi et al., 2015). Arc mRNA increased within 30 min, peaked at 6 h, and remained elevated at 24-48 h. Ketamine also upregulated Klf6 mRNA (peak at 6 h). mRNA levels of PSD95, GluR1, and synapsin I were increased, as was acetylation of H3 and H4 histones. HDAC5 knockdown in DG granule cells partially attenuated ketamine’s antidepressant-like effects; it remained effective in the FST and LH tests, but it lost its efficacy in the NSFT and SPT.
CUS exposure caused depressive behavior, increased HDAC5 in the DG, and reduced HDAC5 phosphorylation; ketamine attenuated every change (Choi et al., 2015). Ketamine-induced downregulation of HDAC5 was observed in CUS animals, but not in control animals, and CUS rats were more reactive to ketamine. HDAC5 knockdown in stressed rats caused antidepressant-like effects, occluding potential benefits from ketamine.
7.16 Other
Sigma Receptors
Opposing Research
Ketamine has negligible affinity for sigma (σ) receptors in the context of its use for depression. Its affinity (Ki) was only 139.60 μM for σ1 and 26.30 μM for σ2 in binding assays using rat liver homogenates in which pentazocine labeled σ1 and DTG labeled σ2; because DTG is not selective between the receptors, the assay was run in the presence of pentazocine to block σ1 (Robson et al., 2012). For comparison, imipramine had a drastically higher affinity of ~300 nM for both σ receptors. When combined with a subeffective concentration of NGF in PC12 cells, imipramine increased neurite outgrowth at 0.1-10 nM, but it was ineffective at 10 to 10,000 nM; ketamine increased outgrowth at all concentrations from 0.01 to 10,000 nM. The σ1 antagonist NE-100 reduced the potentiation of neurite outgrowth induced by ketamine (100 nM) and imipramine.
FST immobility in male mice was reduced by ketamine at 40 mg/kg (IP), with nonsignificant reductions at 10-20 mg/kg; it only had a significant effect at 30 min, not 24-72 h (Robson et al., 2012). The σ1 antagonists NE-100 and BD1047 did not affect immobility on their own nor did they attenuate ketamine’s effect.
MAPK
Supporting Research
MAPK kinase (MAPKK; MEK) is an upstream regulator of proteins in the mitogen-activated protein kinase (MAPK) family. MEK inhibition with PD184161 attenuated ketamine’s antidepressant-like effect in male rats, although MEK inhibition on its own also reduced FST immobility. Ketamine (15 mg/kg IP) was administered three times before testing at 30 min, 60 min, and 1 day before (Reus et al., 2014). MEK inhibition attenuated the effect of ketamine on ERK1/2, p38MAPK, and proBDNF. Phosphorylation of ERK1/2 was reduced in the amygdala, hippocampus, and PFC when the inhibitor was given with ketamine compared to ketamine-only; p-ERK1/2 in the NAc was reduced in every group.
Ketamine increased ERK1 in the PFC, which persisted when the inhibitor was added (Reus et al., 2014). Both ketamine groups had increased ERK1 in the hippocampus compared to saline-treated animals. In the amygdala and PFC, ERK2 was increased in every treatment group compared to vehicle; ERK2 in the NAc was reduced by MEK inhibition and when the inhibitor was used with ketamine, but not when ketamine was used alone. Hippocampal ERK2 was only significantly reduced in the ketamine-only group, with MEK inhibition partially attenuating the effect.
The MEK inhibitor reduced p38MAPK in the amygdala and NAc, with ketamine partially attenuating those changes; in contrast, p38MAPK was increased in the PFC in every treatment group (Reus et al., 2014). proBDNF was only significantly increased in the amygdala in the inhibitor + ketamine group. Only ketamine increased proBDNF in the NAc, while it was reduced in both inhibitor groups. In the PFC, the inhibitor nonsignificantly reduced proBDNF and there were significant reductions in both ketamine groups. Hippocampal proBDNF was increased by ketamine and MEK inhibition attenuated that effect.
Potassium Channels
Kir4.1 are inwardly-rectifying K+ channels that regulate the resting membrane potential (RMP) of astrocytes. Overexpression may be associated with MDD, but the evidence is weak and there is little reason to think it is a good target for antidepressants. R-ketamine does not seem to work through Kir4.1 channels (Xiong et al., 2019).
Opposing Research
Kir4.1 expression in the PFC, hippocampus, and NAc was not affected by R-ketamine (10 mg/kg IP), sertraline, or quinacrine in CSDS-susceptible mice; sertraline and quinacrine are Kir4.1 inhibitors (Xiong et al., 2019). Without affecting Kir4.1, R-ketamine produced rapid and persistent antidepressant effects in susceptible mice in the TST, FST, and SPT. Sertraline and quinacrine were ineffective.
Relevant Non-Ketamine Research
Humans
Xiong et al. (2019) studied postmortem brain samples (cerebellum and parietal cortex) from MDD (n=15), BD (n=15), schizophrenia (n=15), and control (n=15) subjects. In MDD subjects, but not in BD and schizophrenia subjects, Kir4.1 expression in the parietal cortex was increased. MDD and schizophrenia patients also had elevated GABAB receptor subunit 1 levels in the parietal cortex. There were no changes in the cerebellum with any disorder.
Animals
Depressive behavior in congenitally learned helpless (cLH) rats is associated with upregulated astroglial Kir4.1 in the lateral habenula (LHb), a region where increased bursting activity may contribute to depression (Cui et al., 2018). Kir4.1 expression positively correlated with astroglial membrane hyperpolarization and bursting activity. The depression-related increase in astroglial Kir4.1 did not seem to involve astrogliosis (increase in astrocytes following CNS damage) since GFAP was not increased. LPS-induced depressive behavior was also associated with increased Kir4.1 protein and mRNA in the LHb, indicating some of the change could involve transcription regulation. Furthermore, depressive behavior was induced by Kir4.1 upregulation using genetic manipulation.
Calcium Channels
Opposing Research
Antidepressant effects in the FST and SPT were observed with R-ketamine (10 mg/kg IP) administered to CSDS-susceptible mice, but the T-type voltage-dependent calcium channel (T-VDCC) blocker ethosuximide was ineffective in those behavioral tests (Tian et al., 2018).
Section 8: The Role of Metabolism
Appreciable levels of multiple ketamine metabolites are produced by antidepressant doses and some have pharmacological profiles that could be relevant to ketamine’s effect. One metabolite in particular, (2R,6R)-HNK, which is produced from R-ketamine, has received a lot of attention. After research was published showing it has ketamine-like antidepressant affects in animals without causing NMDAR antagonism and that blocking ketamine’s metabolism to (2R,6R)-HNK can inhibit ketamine’s efficacy, some researchers began to suggest (2R,6R)-HNK is vital for ketamine’s effects. If it is, that would further support the importance of R-ketamine over S-ketamine. As more research has been published and old research has been re-examined, it has become clear that (2R,6R)-HNK is at most a contributing factor to the antidepressant effects and that ketamine—potentially along with some of its other metabolites—is highly important.
In addition to (2R,6R)-HNK, there is a small amount of research suggesting norketamine has relevant properties.
8.1 (2R,6R)-HNK
(2R,6R)-hydroxynorketamine, aka (2R,6R)-HNK, is a metabolite of R-ketamine that has antidepressant-like effects in animals. Although it may contribute to ketamine’s mechanism, it is unlikely to be required considering the efficacy of S-ketamine and the efficacy of ketamine locally injected into the brain, which yields far smaller concentrations of (2R,6R)-HNK. Furthermore, some studies have reported it is inactive at relevant doses or less potent than ketamine. Overall, the research suggests that when ketamine or R-ketamine is used, this metabolite contributes to the observed effects but is not vital.
Supporting Research
Animals
The first study to propose a major contribution of (2R,6R)-HNK to the effects of ketamine was published by Zanos and colleagues in 2016. They initially observed that ketamine was more potent in female mice undergoing the FST, prompting them to question whether that difference could involve differential brain penetration of ketamine and its metabolites between the sexes (Zanos et al., 2016). Ketamine and norketamine were found at equivalent levels in the brains of female and male animals, but female mice had a 3-fold higher concentration of (2S,6S;2R,6R)-HNK.
Zanos et al. deuterated ketamine at C6 to study its effects in the relative absence of metabolism since 6,6-dideuteroketamine did not significantly differ from ketamine in terms of NMDAR affinity or brain accumulation, but metabolism was impaired. Deuterated ketamine (10 mg/kg IP) was ineffective in the FST and LH tests at 24 h, though it still reduced immobility at 1 h. The HNK metabolites that were more concentrated in female mice were then tested on their own. (2R,6R)-HNK (10-20 mg/kg IP) was more potent in behavioral tests at 24 h, but (2S,6S)-HNK was still active; (2R,6R)-HNK remained effective on Day 3 and it attenuated social avoidance induced by CSDS and anhedonia (evaluated using SPT and FUST) induced by chronic corticosterone exposure.
(2R,6R)-HNK produced its effects without altering NMDAR function (Zanos et al., 2016). It did not displace MK-801 at NMDAR in vitro nor did it functionally inhibit NMDARs expressed on interneurons in hippocampal slices, whereas it increased AMPAR-mediated EPSPs in the CA1 region of hippocampal slices following stimulation of Schaffer collateral axons; consistent with that effect, it increased the frequency and amplitude of AMPAR-mediated EPSCs recorded from CA1 stratum radiatum interneurons receiving glutamatergic input from Schaffer collaterals. NBQX blocked its efficacy in the FST at 1 and 24 h, indicating AMPAR is functionally important for (2R,6R)-HNK.
As with ketamine, (2R,6R)-HNK increased γ power, indicative of increased activation of fast ionotropic excitatory receptors (e.g. AMPARs), which was attenuated by NBQX (Zanos et al., 2016). Other mechanisms relevant to ketamine’s efficacy were also implicated in the metabolite’s effect, including mTORC1 activation, elevated BDNF, and reduced eEF2 phosphorylation. Neither ketamine nor (2R,6R)-HNK affected p-mTOR levels in mouse hippocampal and PFC synaptoneurosomes, but at 10 mg/kg in vivo, both reduced hippocampal eEF2 phosphorylation at 1 and 24 h, increased hippocampal BDNF at 24 h, and increased hippocampal GluR1 and GluR2 at 24 h. When given 30 min before behavioral testing, NBQX blocked the reduction in FST immobility induced by both drugs.
Despite sharing antidepressant-relevant effects, (2R,6R)-HNK had a lower side effect burden than ketamine (Zanos et al., 2016). It did not increase locomotor activity or cause motor incoordination (rotarod test), whereas ketamine and (2S,6S)-HNK did. Sensory gating (assessed by PPI and startle amplitude) was impaired by ketamine, but not by (2R,6R)-HNK at up to 375 mg/kg. It did not produce ketamine-related discrimination responses in ketamine-treated rats, but PCP did, and it also did not elicit IV self-administration, unlike ketamine. Like ketamine, the circulating concentration of (2R,6R)-HNK quickly peaked around 10 min after IP administration and was greatly reduced within four hours.
In male mice, (2R,6R)-HNK (10 mg/kg IP) and ketamine reduced FST immobility at 24 h and increased mPFC extracellular serotonin (Pham et al., 2017). Basal glutamate release was similarly enhanced in the mPFC by (2R,6R)-HNK, which had a greater effect than ketamine; neither drug appeared to reduce glutamate uptake and instead caused pyramidal cell glutamate release. The systemic effects were replicated when either drug was bilaterally infused into the mPFC. Intra-mPFC infusion of ketamine also increased extracellular GABA, but (2R,6R)-HNK did not.
A single dose of (2R,6R)-HNK (10 mg/kg IP) reversed LH-induced depressive behavior in the social avoidance test within one hour and its behavioral effects were maintained for up to 21 days (FST and SPT) in male and female rats (Chou et al., 2018). The same dose of (2S,6S)-HNK was inactive in the FST and SPT. Stress-induced reductions in glutamatergic transmission and GluR1 expression in midbrain ventrolateral periaqueductal gray (vlPAG) neurons were reversed by (2R,6R)-HNK. In vitro, (2R,6R)-HNK (10 μM) increased mEPSC frequency and amplitude in vlPAG neurons, and intra-vlPAG administration in vivo caused rapid and long-lasting antidepressant-like effects, which were blocked by AMPAR antagonism with intra-vlPAG CNQX. The metabolite was similarly effective against depressive behavior caused by chronic restraint stress or spinal nerve ligation. There was no difference between male and female rats at 1 or 24 h.
The oral bioavailability of (2R,6R)-HNK is superior to that of ketamine: 46-52% in mice, 42% in rats, and 58% in dogs (Highland et al., 2019). It also readily penetrates the brain, producing brain:plasma ratios of 0.67-1.2 in mice and rats. Oral administration (15-150 mg/kg) reduced FST immobility at 1 h and 24 h, as did systemic ketamine (15 mg/kg IP) and (2R,6R)-HNK (15 mg/kg IP). It reduced escape failures in the LH paradigm at 50 mg/kg (IP) and when given orally at 50 and 150 mg/kg.
BDNF and mTORC1 may be involved in the antidepressant mechanism of (2R,6R)-HNK. Mice with the BDNF Val66Met allele and mice exposed to an intra-mPFC injection of an anti-BDNF neutralizing antibody did not respond to 30 mg/kg (IP) of the drug (Fukumoto et al., 2019). Antagonizing L-type VDCCs (with verapamil), which are required for activity-dependent BDNF release, also blocked the effects. (2R,6R)-HNK increased mPFC synaptic function, with an increase in orexin-induced EPSCs in layer V pyramidal neurons 24 h after dosing and there was a trend-level increase in serotonin-induced currents; this was not associated with changes in total spine density or spine subtypes (stubby, thin, mushroom). TrkB inhibition with ANA-12 blocked the effects of (2R,6R)-HNK.
The role of Rac1, a GTPase that is important for the synaptic effects of BDNF-TrkB signaling, was studied using the Rac1 inhibitor NSC 23766. Intra-mPFC administration of the Rac1 inhibitor attenuated the FST and NSFT effects of ketamine and (2R,6R)-HNK (Fukumoto et al., 2019). At 10 ng/side, (2R,6R)-HNK administered directly to the mPFC had antidepressant effects in the FST and NSFT; ketamine and (2R,6R)-HNK transiently increased p-mTOR at 30 min but not 60 min and intra-mPFC rapamycin blocked the behavioral effects.
(2R,6R)-HNK (30 mg/kg IP) was effective in the FUST one day after dosing, but unlike ketamine (10 mg/kg IP), was ineffective by Day 4; however, both were effective in the FST on Day 5 (Fukumoto et al., 2019). In a primary cortical culture, p-ERK was increased by (2R,6R)-HNK at 1-50 nM, with a reduction at 100-500 nM; that effect was blocked by the TrkB inhibitor K252a and by NBQX. (2R,6R)-HNK increased BDNF release at 10 and 50 nM, which was attenuated by NBQX and the VDCC blocker verapamil; it also increased mTORC1 signaling (increased p-p70S6K), an effect blocked by TrkB inhibition. This suggests its effects involve mTORC1 activity downstream of BDNF-TrkB signaling.
A more recent study from Zanos and colleagues supported their earlier work on (2R,6R)-HNK, but instead of framing the metabolite as vital for ketamine’s efficacy, they argued it contributes to the effects. In male mice, R-ketamine (10 mg/kg IP) and 6,6-dideutero-R-ketamine produced the same NMDAR antagonism, but deuteration inhibited ketamine’s metabolism to (2R,6R)-HNK; the brain Cmax of the metabolite was 4.5-fold lower and its AUC was 7-fold lower. R-ketamine was more potent than deuterated R-ketamine in behavioral tests, supporting an effect of metabolism. FST immobility was reduced at 24 h using 5 or 10 mg/kg R-ketamine, but 10 mg/kg was necessary for R-d2-ketamine.
The same difference was present for depressive behavior in the SPT and escapable shock test after inescapable shock exposure (Zanos et al., 2019). In the SPT, R-ketamine increased sucrose preference at 48 h with 2.5-10 mg/kg, but at least 5 mg/kg was required for R-d2-ketamine; 5 mg/kg R-ketamine was also stronger than 5 mg/kg R-d2-ketamine. Although it was more potent, the effect of R-ketamine did not differ from R-d2-ketamine when they were both given at 10 mg/kg. Five days after exposure, R-ketamine (5 and 10 mg/kg) reduced escape failures, while R-d2-ketamine was only active 10 mg/kg; the magnitude of effect at 10 mg/kg was quantitatively larger with R-ketamine.
ICV infusion of (2R,6R)-HNK dose-dependently produced antidepressant effects, with a reduction in escape failures at 3 and 10 nmol, but no activity at 30 nmol and only trend-level efficacy at 1 nmol (Zanos et al., 2019). In the SPT, ICV administration of (2R,6R)-HNK increased sucrose preference at 48 h with every dose. R-ketamine caused locomotor stimulation, PPI deficits, motor incoordination, and CPP with around half the potency of racemic ketamine. Typically, the required dose (often >40 mg/kg) for these side effects was higher than what is necessary for antidepressant effects.
Lumsden et al. (2019) reported a reduction in FST immobility 1 h and 24 h after (2R,6R)-HNK (10 mg/kg IP), and it reduced latency to feed in the NSFT at 30 min in male mice. Hippocampal slices obtained 30 min after drug exposure showed an increase in p-mTOR and BDNF, without any changes to mTOR or proBDNF. At this dose, the hippocampal extracellular concentration reached a peak of 7.57 μM at 10 min, falling to 2.96 μM at 30 min, 0.37 μM at 1 h, and 0.054 μM at 2 h; therefore, antidepressant effects are obtained with less than 10 μM.
(2R,6R)-HNK did not prevent NMDA-induced lethality at 10 mg/kg (Lumsden et al., 2019). The ED50 for lethality prevention was 6.4 mg/kg for ketamine, yet 228 mg/kg for (2R,6R)-HNK and 19 mg/kg for (2S,6S)-HNK. Its limited effect at NMDAR was confirmed in vitro. In magnesium-free artificial CSF (aCSF), NMDAR-mediated fEPSPs in the CA1 region of mouse hippocampal slices were reduced by ketamine and the HNK metabolites, but with substantially different potencies. The IC50 of ketamine was 4.5 μM, while it was 212 μM for (2R,6R)-HNK and 47 μM for (2S,6S)-HNK. Ketamine inhibited NMDAR-mediated fEPSPs by 76% at 10 μM and 89% at 100 μM, but 10-100 μM of either HNK enantiomer did not inhibit those responses.
Ketamine more effectively reduced the amplitude of mEPSCs in rat CA1 pyramidal neurons, with an EC50 of 6.4 μM compared to 64 μM for (2R,6R)-HNK (Lumsden et al., 2019). When whole-cell currents in rat CA1 pyramidal neurons were induced by a mixture of NMDA and glycine while in magnesium-containing aCSF, the IC50 for inhibiting the charge carried by NMDAR-mediated currents was 46 μM for ketamine, whereas up to 1,000 μM of (2R,6R)-HNK did not have an effect; the high IC50 of ketamine may be related to the presence of magnesium. Regardless of subunit composition, (2R,6R)-HNK was largely inactive at NMDARs, as shown in Xenopus oocytes expressing recombinant receptors; in this assay, current amplitude was measured after the application of glutamate + glycine.
- Rank order for (2R,6R)-HNK: GluN1/N2C receptors (IC50 = 202 μM) ~ GluN1/N2B (IC50 = 258 μM) ~ GluN1/N2D (IC50 = 287 μM) > GluN1/N2A (IC50 = 498 μM).
- Rank order for (2S,6S)-HNK: GluN1/N2D (IC50 = 13 μM) = GluN1/N2C (IC50 = 15 μM) > GluN1/N2B (IC50 = 21 μM) > GluN1/N2A (IC50 = 43 μM).
Opposing Research
Humans
There was no correlation between (2R,6R)-HNK plasma concentration and antidepressant response at 230 min in MDD (n=45) and BD (n=22) patients treated with ketamine (0.5 mg/kg IV, 40-min) (Zarate et al., 2012). During the first 230 min, ketamine, norketamine, DHNK, four HNKs, and hydroxyketamine (HK) were detected. BD patients had higher DHNK, (2S,6S;2R,6R)-HNK, (2S,6R;2R,6S)-HNK, and (2S,5S;2R,5R)-HNK, whereas MDD patients had higher (2S,6S;2R,6R)-HK. Nonresponse in BD patients was correlated with higher (2S,5S;2R,5R)-HNK.
Animals
Metabolism does not seem to be a necessary component of R-ketamine’s mechanism. In rats exposed to the LH paradigm to induce depressive behavior, a single bilateral infusion of R-ketamine (2 μg/side) produced antidepressant-like effects in the conditioned avoidance test when infused into the IL mPFC, CA3, or DG (Shirayama and Hashimoto, 2016). R-ketamine was not effective when infused into the prelimbic mPFC, NAc shell or core, basolateral amygdala, or central nucleus of the amygdala.
Depressive behavior induced by CSDS or LPS exposure in male mice was not alleviated by (2R,6R)-HNK (10 mg/kg IP), whereas R-ketamine (10 mg/kg) produced rapid, long-lasting effects and was more potent than S-ketamine (Yang et al., 2017). One dose of either ketamine enantiomer reduced depressive behavior (assessed with FST) in the LPS model, but (2R,6R)-HNK only had weak effects. And in CSDS-exposed mice, R- and S-ketamine had rapid antidepressant effects that persisted for at least one week, as assessed in the SPT (Days 3 and 7), TST (Day 1), and FST (Day 2); R-ketamine was more potent than S-ketamine and (2R,6R)-HNK was inactive.
R-ketamine (20 mg/kg IP) was effective in the LH model when conditioned avoidance was tested in male rats 24 h or five days after drug exposure, but R-norketamine (20 mg/kg) and (2R,6R)-HNK (20 or 40 mg/kg) were not (Shirayama and Hashimoto, 2018).
Zhang et al. (2018) tested the effects of ICV administration of R-ketamine and (2R,6R)-HNK in the CSDS model. R-ketamine had rapid and long-lasting effects for at least one week, but (2R,6R)-HNK was ineffective in the TST, FST, and SPT despite its brain concentration being larger when it was administered directly compared to the amount detected after R-ketamine infusion—the (2R,6R)-HNK detected in the brain after ICV administration of R-ketamine likely came from peripheral metabolism following removal of R-ketamine from the brain; the metabolite was present in the liver and blood one hour after ICV infusion of either R-ketamine or (2R,6R)-HNK.
When C6 was deuterated, R-ketamine (10 mg/kg IP) continued to show rapid, long-lasting (7 day) effects in the mouse CSDS model even though deuteration reduced metabolism to (2R,6R)-HNK, whereas it did not affect plasma and brain levels of R-ketamine or R-norketamine (Zhang et al., 2018).
Attenuation of R-ketamine metabolism to (2R,6R)-HNK in mice using CYP450 inhibitors (ticlopidine and 1-aminobenzotriazole) enabled a small subtherapeutic dose of R-ketamine (3 mg/kg IP) to become active in the LPS model (Yamaguchi et al., 2018). The inhibitors greatly reduced (2R,6R)-HNK production, although they also increased the blood AUC of R-ketamine (10 mg/kg) by 10x and the Cmax by 3x, complicating the interpretation of these results. R-norketamine (10 mg/kg) and (2R,6R)-HNK (10 mg/kg) were ineffective in the LPS model, as assessed by FST behavior. The increased potency of R-ketamine in the presence of enzyme inhibitors is not a good indicator of how effective R-ketamine is without (2R,6R)-HNK since the inhibitors also substantially increased brain levels of R-ketamine; this could mask what would normally be a reduction in activity if (2R,6R)-HNK was lost without a concurrent increase in R-ketamine.
Relevant Research
Animals
LTP in the NAc was impaired in mice 24 h and one week after ketamine (3 and 10 mg/kg IP) or (2R,6R)-HNK (10 mg/kg) (Yao et al., 2017). Although it inhibited LTP on its own, rapamycin did not block the ketamine-induced reduction in LTP. Ketamine attenuated an increase in LTP-induced phosphorylation of GluR1 at the CaMKII/PKC site (Ser831), while causing a persistent increase in GluR1 phosphorylation at the PKA site (Ser845). AMPAR-mediated responses were reduced in VTA dopaminergic neurons 24 h after ketamine or (2R,6R)-HNK without an effect on NMDAR-mediated responses, indicative of LTD. This suggests both drugs reduce LTP in the NAc and induce LTD at these glutamatergic synapses.
In Vitro
Suzuki et al. (2017) confirmed that (2R,6R)-HNK did not inhibit NMDARs in cultured hippocampal neurons at concentrations likely to be reached with ketamine for depression. It did not affect NMDAR-mediated charge transfer at 10 μM, though it caused a 40% reduction at 50 μM. When applied alongside TTX, CNQX, and picrotoxin in the absence of magnesium to isolate and enhance NMDAR-mediated transmission, (2R,6R)-HNK (50 μM) produced a shift towards smaller NMDAR-mEPSC amplitude, indicating a postsynaptic effect. Likely contradicting the results of Zanos et al. (2016), a larger concentration of the metabolite (50 μM, not 10 μM) was required to decrease p-eEF2.
Ketamine increased dendritic arborization in dopaminergic neurons at ~1 μM, along with increasing soma size in mouse mesencephalic cultures and human iPSC-derived dopaminergic neurons (Cavalleri et al., 2018). Activation of mTORC1 signaling was shown by an increase in p-p70S6K; ketamine’s effects on mTORC1 and dendritic arborization were attenuated by rapamycin and the PI3K inhibitor LY294002. At sub-micromolar concentrations, (2R,6R)-HNK mimicked ketamine’s effects, increasing structural plasticity in vitro at 0.5 μM and at a trend-level with 0.1 μM.
The effects of ketamine depended on intact AMPAR, BDNF, and dopamine receptor activity (Cavalleri et al., 2018). AMPAR antagonists attenuated ketamine’s effect, whereas ketamine-like changes were produced by the AMPAR PAM CX614; inhibiting BDNF signaling prevented ketamine-induced increases in structural plasticity; and the effects were also blocked in D3 receptor knockout cultures and when D3 antagonists were used.
In an in vitro study using human iPSC-derived dopaminergic neurons, submicromolar (2R,6R)-HNK exposure for 6 h increased dendrite outgrowth when measured three days later; its effect was like that of low-micromolar ketamine exposure for 1 or 6 h (Collo et al., 2018). Both drugs were blocked by AMPAR antagonism (NBQX or GYKI 52466) and by rapamycin. (2R,6R)-HNK was more potent, with its activity at 0.5 μM being roughly similar to 1 μM of ketamine.
8.2 Norketamine
Compared to ketamine and (2R,6R)-HNK, few studies have looked at the effects of norketamine, but a small amount of evidence suggests it could be an antidepressant (Hashimoto and Yang, 2019). In particular, Yang et al. (2018) found it had antidepressant-like effects akin to ketamine in rodents. Its efficacy seemed to involve an AMPAR-independent mechanism that was dependent on mTORC1 and BDNF signaling. Norketamine produced its antidepressant effects in rodents without the major side effects of ketamine.
Supporting Research
Humans
Norketamine concentration at 90 min negatively correlated with depressive symptoms at 24 h in 11 MDD patients treated with ketamine (0.5 mg/kg IV, 40-min) (Milak et al., 2016). In contrast, ketamine and DHNK concentration did not correlate with clinical outcome.
Animals
In the LPS and CSDS models of depression, S-norketamine (5-10 mg/kg IP) had more potent antidepressant-like effects than R-norketamine (Yang et al., 2018). S-norketamine was effective at 5-10 mg/kg in the LPS model, while R-norketamine required 20 mg/kg. Similarly, S-norketamine was stronger than R-norketamine at 10 mg/kg in the CSDS model and the benefits were long-lasting, with efficacy persisting for at least four days. Only S-norketamine attenuated CSDS-induced TST and FST immobility one day after drug exposure; it also increased sucrose preference one week after treatment. The superior behavioral effects of S-norketamine were associated with greater attenuation of CSDS-induced reductions in dendritic spine density and synaptogenesis in the PFC and hippocampus.
S-norketamine appeared to work through an AMPAR-independent mechanism since AMPAR antagonism (with NBQX or CNQX) did not block its effects (Yang et al., 2018). Electrophysiological experiments found S-norketamine antagonized NMDAR-mediated currents without enhancing AMPAR-mediated transmission in hippocampal neurons. TrkB antagonism (with ANA-12) and mTORC1 antagonism (with rapamycin) blocked S-norketamine. Out of 80 receptors, ion channels, and transporters, NMDAR was the only target inhibited by >50% with 10 μM S-norketamine.
Unlike S-ketamine (10 mg/kg), S-norketamine did not cause sensorimotor gating deficits (PPI), rewarding effects, loss of PV immunoreactivity in the mPFC, or increased γ oscillations at baseline (Yang et al., 2018).
Opposing Research
Humans
There was no difference between responders and nonresponders in plasma ketamine or norketamine at 10 and 30 min, although plasma norketamine at 10 min correlated with BPRS-Anergia score (i.e. emotional withdrawal, motor retardation, blunted affect, and disorientation) in a DBRCT crossover study of 27 hospitalized depression patients (Sos et al., 2012).
Ketamine and norketamine levels also did not correlate with depression reduction or acute dissociation among 108 treatment-resistant depression patients (MDD=74; BD=34) (Luckenbaugh et al., 2014).
Section 9: Risks
Ketamine has been studied in thousands of healthy people, surgical patients, and psychiatric patients through research on its analgesic, anesthetic, antidepressant, anxiolytic, and psychotomimetic effects. Those studies have demonstrated it is largely free of serious acute risks, but it commonly causes short-lasting side effects, particularly psychological and perceptual changes (dissociative, psychotomimetic), cognitive impairment, and cardiovascular stimulation (modest BP and HR increase). Those changes most often subside without issue within 2-4 hours. In the event of more serious cases of those acute effects, the effects still tend to subside soon after exposure; termination of an IV infusion usually produces resolution within ~30 minutes without other treatment being required.
The literature likely suffers from underreporting and inadequate analysis of the adverse effects since trials are known to be deficient in that area, partly because adverse effects are nearly always a secondary outcome; therefore, studies are not powered and designed to detect those differences (Zorzela et al., 2014). That said, its acute safety profile is mostly understood due to substantial research and clinical observation. Some of the only remaining questions pertain to how ketamine interacts with specific conditions, such as psychotic disorders. Additionally, while it has become more common to research repeated dosing, most controlled trials are on single doses, so questions remain about its long-term safety in people who are frequently exposed to the drug, including psychiatric and pain patients (Short et al., 2018).
9.1 Acute Risks
Psychological Side Effects
A review of 60 studies covering 899 depression patients reported that the most common acute psychiatric side effects were anxiety, agitation or irritability, euphoria or mood elevation, delusions or unusual thoughts, panic, and apathy (Short et al., 2018). The top psychotomimetic side effects included dissociation, perceptual changes, odd/abnormal sensations, derealization, hallucinations; feeling strange, bizarre, weird, or unreal; and depersonalization. Ketamine was not associated with persistent psychiatric side effects, but most reports only studied patients for a short time after exposure.
In healthy people, a review of 16 studies (450 people; 833 ketamine and 621 placebo infusions) found 10 adverse mental events in nine subjects/infusions, giving a ~2% occurrence, and only one did not resolve by the end of the session; the one extended adverse event mostly subsided within four days (Perry et al., 2007). Ketamine was given intravenously as a bolus + infusion or as a continuous infusion at subanesthetic levels. Some studies involved coadministration with clozapine, haloperidol, amphetamine, glycine, lamotrigine, naltrexone, the mGluR2/3 agonist LY354740, nicotine, amphetamine, or lorazepam. Amphetamine was the only drug that worsened ketamine’s psychotomimetic effects.
Among the nine subjects with adverse mental events, three became unresponsive to verbal stimuli yet quickly returned to normal responsiveness after infusion discontinuation:
- Subject 1: Received oral LY354740 alongside ketamine. She became nonverbal and could squeeze the nurse’s hand on command. Responsiveness returned within 2 min of infusion discontinuation. She reported having felt like she was in a movie and she didn’t think verbal communication was necessary because the staff could read her mind.
- Subject 2: Received oral LY354741 alongside ketamine. He became nonverbal but could open his eyes when his name was called. He was verbal again within 30 min of ending the infusion. He also experienced nightmares, insomnia, and decreased concentration for 3-4 days. Those symptoms did not prevent him from returning to work and they resolved entirely within 2 weeks.
- Subject 3: Male who was recently withdrawn from alcohol. He received ketamine and was only responsive to deep pain stimuli after 18 min of the infusion (0.5 mg/kg, 40-min). Responsiveness returned within 5 min of infusion discontinuation.
A small minority of participants gave long-term follow-up information 1-6 months post-infusion and none reported emotional, psychological, neurological, or cognitive issues (Perry et al., 2007). They did not experience cravings for ketamine or report use outside of the research setting; ketamine was not associated with persistent altered perception, flashbacks, paranoia, or sluggishness.
Ketamine usually does not aggravate suicidality in patients with mild or severe suicidal ideation, but there is a small possibility that this will occur. For example, Vande Voort and colleagues (2016) reported a case of clinical worsening with behavioral outburst and suicidal threat in a patient who was hospitalized with suicidal ideation and treated with ketamine. The incident occurred after the fifth infusion of a two-week six-infusion series and was associated with “external stressors during hospitalization,” so it is unclear what role ketamine played.
Switch to Mania
Ketamine can transiently increase mood and induce psychotomimetic effects, but it does not seem to cause persistent switch to mania in unipolar or bipolar depression patients. Across three trials of 98 patients, some with unipolar depression and others with BD, there were no cases of persistent manic switch despite it transiently producing substantial mania-like effects in some people (Niciu et al., 2013).
Long-Term Effects of Brief Repeated Dosing
Subacute ketamine with up to four subanesthetic doses (0.1-0.5 mg/kg IV bolus) over a two-week period did not produce long-term adverse effects in schizophrenic patients (n=30) compared with patients who did not receive ketamine (n=25) (Lahti et al., 2001). Ketamine was given in a double-blind manner; 25 patients were available for long-term follow-up. It was minimally distressing and no serious adverse events were reported. The ketamine-treated patients were evaluated over an average follow-up period of eight months and they did not differ from control patients in terms of psychopathology, psychiatric care, or antipsychotic dosage.
There was an acute increase in psychotic symptoms at 20 min, including a 30% increase in positive symptoms compared to placebo, but those scores normalized within 90-180 min (Lahti et al., 2001). Two patients had an acutely unpleasant first experience and declined to participate further: one had a 10-min increase in paranoid ideation that quickly resolved without treatment and the other became anxious secondary to lightheadedness and numbness, but they returned to normal without treatment. During the first 24 h after dosing, one person reported increased visual hallucinations and two had an increase in delusions. None of the subacute adverse effects lasted longer than two days or required treatment, and ketamine could not be definitively identified as the cause.
9.2 Chronic Risks
Cognitive Impairment
Humans
The impairing effects of a single dose subside within a few hours. However, when ketamine is taken frequently (particularly at nonmedical doses), greater cognitive impairment can occur and it may persist for some time after the cessation of use. Impairment from chronic high-dose use appears to substantially decline or resolve within weeks or a few months of stopping use in most people, but there is very little research on this point. Some research suggests high-dose recreational use at a frequency of once per week does not greatly affect cognitive performance when tested one week later (Morgan et al., 2009).
Frequent users had impaired spatial working memory, pattern recognition memory, executive function (Stockings of Cambridge test), and category fluency performance, but verbal fluency and prose recall were not impaired; frequent users (n=30) were compared with infrequent users (n=30), non-ketamine polydrug users (n=30), and non-using controls (n=30) (Morgan et al., 2009). Frequent use was defined as >4x per week, infrequent use was at least monthly, and ex-ketamine users had been abstinent for at least one month (12 had previously been frequent users).
Infrequent and ex-users did not differ from the control groups (Morgan et al., 2009). Frequent users had more delusional, dissociative, and schizotypal symptoms, which were present to a lesser degree in infrequent and ex-users; delusional symptoms positively correlated with the amount of ketamine use reported by frequent users. Frequent users had last taken ketamine 2 days before testing compared with 11 days for infrequent users, and they were currently using it 20 days per month versus 3 days per month in the infrequent group. The duration of regular use was 5 years in frequent users and 3.7 years in infrequent users. Each group had initially taken ~0.5 g per session, but that increased to 3.8 g in frequent users and 1.3 g in infrequent users.
Morgan and colleagues (2010) also studied the effects of ketamine over time in the same participants. After their initial evaluation, 80% were available for a 12-month follow-up. Cognitive deficits were most common in frequent users and their deficits in spatial working memory and pattern recognition memory persisted over time, with trends towards impaired verbal recognition memory. Declines in spatial working memory and pattern recognition memory task performance at follow-up correlated with increased use during the follow-up period; frequent users also had greater dissociative symptoms. Although reported ketamine use did not increase during the follow-up period, the average ketamine concentration in hair samples doubled.
Tang et al. (2013) reported cognitive impairment in current ketamine users (n=51) compared to ex-users (n=49) and healthy controls (n=100). The mean time since last use was 3 days for current users and 189 days for ex-users; current users reported taking ketamine for 5.3 years, while ex-users had taken it for 4.7 years. Current users reported ~17 days of use per month on average. Interestingly, current users had much higher depression scores, with a BDI of 21 (moderate depression) compared to 12 for ex-users (minimal symptoms), and there was a modest correlation between cognitive impairment and depressive symptoms. Impairment was most notable in the domains of mental and motor speed, visual and verbal memory, and executive function.
Animals
Mice treated with ketamine (20 mg/kg/d IP) for 14 days had a reduction in amplitude for components of event-related potentials and reduced stimulus-evoked theta oscillations six months later (Featherstone et al., 2012). Ketamine also caused persistent impairments in reversal learning and spatial memory. There was no sign of neuronal degeneration (using NeuN immunohistochemistry), but ketamine-treated mice had an increase in astrocyte proliferation and reduced expression of the glutamate transporter GLT-1.
Urological Toxicity
Though it is far from universal, a common adverse effect of heavy chronic use is urological toxicity, presenting with symptoms like greatly increased urination urgency and frequency, dysuria (pain when urinating), nocturia (excessive nighttime urination), lower abdomen/suprapubic pain, and occasionally hematuria (blood in urine). Those symptoms are often associated with bladder inflammation (cystitis).
At least one urinary symptom occurred in 27% (n=340) of people who reported past-year ketamine use (n=1285) in a survey of 3806 people (Winstock et al., 2012). They were asked whether they had experienced lower abdomen pain, burning or stinging during urination, increased urinary frequency, incontinence, and/or hematuria. Symptoms were more common in people who used ketamine at higher doses and with greater frequency. 51% experienced an improvement in symptoms following cessation, but the symptoms remained constant post-discontinuation in 43%, and worsened in 4%.
Average per session use for ketamine users overall was: <0.125 g (31%), 0.25-0.5 g (35%), and ≥1 g (34%) (Winstock et al., 2012). The most they had taken in a session was >1 g (54%), >3 g (25%), or >6 g (7%). On average, the max number of consecutive days of use was 3.5, with 12% of users having previously taken it ≥7 days in a row. 792 (62%) had taken ketamine in the past month and they reported using it an average of four times per month. 17% of users met the criteria for dependence.
Middela and Pearce (2010) reviewed 20 reports with 111 patients who experienced urological symptoms associated with ketamine; the cases were highly variable. Onset of lower urinary tract symptoms (LUTS) occurred a few days to a few years after initiating abusive ketamine use, with some users taking grams each day. Patients typically had irritative symptoms (urgency, frequency, dysuria) and there was no reliable treatment. Cessation of use appeared to be the best option, whereas antibiotics, NSAIDs, steroids, anticholinergics, and cystodistension did not provide persistent benefit. The more severe cases involved greatly reduced bladder capacity and compliance, cystitis, and hydronephrosis (kidney swelling due to urine buildup).
In nine chronic ketamine users who were experiencing severe urological symptoms, CT scan showed inflammation-indicative changes, including bladder wall thickening and reduced capacity, and cystoscopy revealed ulcerative cystitis (Shahani et al., 2007). Their symptoms began months before presentation and after the initiation of daily or near-daily use. Abstinence usually reduced the symptoms.
Cystitis was reported in an adolescent female who received daily oral ketamine for the treatment of complex regional pain syndrome following failure with other medications (Gregoire et al., 2008). Her dose reached 8 mg/kg/d shortly after initiation and by Day 9 she had dysuria, frequency, urgency, and incontinence. Dose reduction to 6 mg/kg/d partially attenuated the symptoms and they fully resolved at 2 mg/kg/d. After a period in which ketamine was not used, it was restarted at 5 mg/kg/d, producing reappearance of urological symptoms; dose reduction to 3 mg/kg/d resolved the issue.
A retrospective chart review of 59 ketamine users who presented to hospitals in Hong Kong with severe LUTS found the most common symptoms were frequency, urgency, dysuria, urge incontinence, and less often painful hematuria (Chu et al., 2008). The mean duration of use was 3.5 years. Cystoscopy was performed in 71%, which showed epithelial inflammation in the bladder, and bladder biopsies (n=12) found histological changes akin to interstitial cystitis. Renal ultrasonography showed unilateral or bilateral hydronephrosis in 51% and 7% had kidney damage, specifically papillary necrosis. Creatinine was elevated in eight patients.
Shortly after initiating twice-weekly intranasal use, a 21-year-old male began experiencing LUTS and he presented nine months later with frequent urination, nocturia, and suprapubic pain; microscopic hematuria was observed on urine analysis (Ho et al., 2009). Cystoscopy showed inflammation of the bladder mucosa consistent with cystitis. Cessation of use resolved the symptoms and he was asymptomatic three months later.
Daily oral ketamine prescribed for pain caused urological symptoms in three patients (Storr and Quibell, 2009). In the first patient, symptoms began after her dose reached 50 mg (oral) four times per day (qds), with hematuria, dysuria, and perineal pain; cystoscopy showed contracted bladder and inflammatory cell infiltration. Her use continued for a year until she was admitted for bladder pain, at which time she was on 200 mg five times per day. Her symptoms resolved within three weeks of discontinuation.
In the second patient, suprapubic pain, urinary frequency, and an episode of hematuria developed five months into treatment with 170 qds; inflammation was seen on cystoscopy (Storr and Quibell, 2009). Because ketamine was not initially identified as the cause, her dose was increased to 200 mg qds for analgesia. Five months later, she was admitted due to exhaustion and she was urinating hourly, had substantial bladder pain, and her urine white blood cell count was elevated (>500). Bladder pain resolved a month after cessation, but increased urinary frequency and nocturia continued until her death 18 months later.
A third patient developed hematuria five months into treatment with 130 mg qds and one month later he was admitted with suprapubic pain and substantial hematuria necessitating blood transfusion; cystoscopy showed cystitis (Storr and Quibell, 2009). The dose was increased to 200 mg five times daily for pain relief. Four weeks later, he was urinating up to 12 times each night and had severe bladder pain; both issues resolved after cessation.
Cystitis accompanied by urinary frequency and urgency, but not hematuria, dysuria, or pain, was reported in a female who had initiated recreational ketamine use one year earlier (Noorzurani et al., 2010). After 2-3 months she began taking it daily because of dependency; she also reported binge alcohol use each weekend. Ultrasound showed normal kidneys. Her urinary frequency increased to 2-3x per hour and she had to wear diapers during sleep. Despite being advised to discontinue ketamine use, she continued taking it at a reduced frequency and her symptoms persisted.
Tam et al. (2014) studied the effects of chronic ketamine use among active users (n=214) and ex-users (n=104) in Hong Kong who reported urological symptoms. Ex-users had been abstinent for at least a month (median = 4 months). All users had to have taken ketamine for at least six months with a frequency of ≥2x per month and the inclusion criteria required the presence of two or more LUTS in the past three months, such as hematuria, dysuria, urinary frequency, urgency, nocturia, or suprapubic/pelvic/perineal pain. The average duration of use was 81 months, with an average of 18.5 g taken per week. Although 50% engaged in polydrug use, none reported regular use of drugs other than ketamine. All users took ketamine intranasally.
The median duration of LUTS was 24 months and there were reductions in voided volume (mean: 112 mL) and bladder capacity (mean: 153 mL); the mean bladder emptying efficiency was 73% (Tam et al., 2014). Renal impairment (creatinine >90 μmol/L) was present in 7%. Ultrasound examination was performed in 160 patients, which revealed bladder wall thickening (92%) and unilateral or bilateral hydronephrosis (8%). Ex-users had a larger average voided volume (126 vs. 85 mL) and bladder capacity (205 vs. 127 mL). Female sex was associated with more severe symptoms and smaller voided volume, and magnitude of ketamine use was also associated with symptom severity.
Gastrointestinal Toxicity
Chronic use has been associated with abdominal pain (”k-cramps”) in some nonmedical users. A retrospective study found upper gastrointestinal (GI) symptoms were present in 28/37 users receiving treatment for ketamine-related issues (Poon et al., 2010). 82% only had epigastric pain, while 14% had pain with vomiting. In the total sample, 90% (33/37) had urological symptoms. Upper GI endoscopy was performed in 14 patients, which revealed stomach inflammation (n=12), stomach and duodenum inflammation (n=1), and nothing abnormal (n=1). The mean duration of ketamine use was four years.
Tolerance
Few studies have administered ketamine long enough to properly evaluate whether tolerance develops to its effects in the context of psychiatric use beyond a few months. When it is used at large doses in nonmedical settings, tolerance readily develops to its dissociative, euphoric, hallucinogenic, and psychotomimetic effects. Its antidepressant effect may also shorten in duration if overused (Bonnet, 2015).
9.3 Neurotoxicity
Human Research
People dependent on ketamine (n=41) had reduced gray matter volume in the bilateral frontal cortex compared with non-users (n=44) (Liao et al., 2011). Ketamine users were recruited from drug rehab centers and they were excluded if they were dependent on anything other than ketamine and nicotine; they were also excluded if they had a major medical/psychiatric disorder or were using psychiatric medications. The mean duration of use was 41 months (range: 12-126 months) and ketamine users reported a significant lifetime history of other drug intake, including tobacco (n=41), alcohol (n=30), ecstasy (n=28), amphetamine with caffeine (n=27), methamphetamine (n=27), cannabis (n=8), and benzodiazepines (n=6).
All users took ketamine intranasally; four reported bladder dysfunction and seven had chronic gastritis, issues that were absent in control subjects (Liao et al., 2011). Structural MRIs were obtained after ≥48 h of abstinence. Longer duration of use and higher lifetime intake correlated with smaller gray matter volume, specifically in the left superior frontal gyrus. Gray matter volume did not correlate with age, years of education, or age at initiation of ketamine use. Users exhibited deficits in cognitive function, as evaluated in the Digit Symbol Test and Stroop Color-Word Test. Most had experienced schizophrenia-like effects from their use and 23 reported anxiety and/or depressive experiences; gray matter volume did not correlate with those effects.
Atrophy in the frontal, parietal, and occipital cortices was observed on MRI in ketamine-dependent people (n=21) compared with control subjects (n=3) (Wang et al., 2013). The ketamine-associated lesions positively correlated with duration of use, often appearing after 2-4 years of regular use (Wang et al., 2013). 19/21 took ketamine daily, the average daily intake was 1 g (range: 0.2-3 g), and duration of use ranged from 0.5 to 12 years: <3 years (n=6), 4-6 years (n=7), 7 years (n=3), and >10 years (n=5). All users took ketamine intranasally.
The initial abnormalities, which could be seen within one year of initiating use in some cases, were hyperintense regions potentially associated with cortical white matter degeneration; those abnormalities spread to the internal capsule within three years and were present in the basal forebrain, cerebellum, pons, and diencephalon in 3-4 years (Wang et al., 2013). Atrophy in the parahippocampal gyrus was seen by five years and cortical atrophy began within four years, spreading from a small area to 2-3 regions within seven years of use. Hyperintense regions were observed in the corpus striatum within six years. Those who had taken ketamine for more than four years had memory impairment along with anxiety and/or depressive symptoms; ataxia was observed in people with 5-7 years of use and dyskinesia occurred at >7 years. A major caveat to this progression timeline is that the sample size for each use-duration group was very small, limiting the generalizability and reliability of the results.
Animal Research
In 1989, Olney and colleagues published a study demonstrating that NMDAR antagonists (MK-801, PCP, and ketamine) produce neurotoxicity in the form of vacuoles in the retrosplenial cortex of rats, which are now known as Olney’s lesions (Olney et al., 1989). Affinity for NMDAR correlated with neurotoxic potency. The vacuoles formed 4 h after a single dose of ketamine at 40 mg/kg (SC), but not at 10-20 mg/kg.
Ketamine and nitrous oxide, both of which are neurotoxic in female rats, synergistically increased toxicity in posterior cingulate/retrosplenial cortex (PC/RSC) neurons when administered together (Jevtovic-Todorovic et al., 2000). The ED50 of ketamine was 47.5 mg/kg (IP), while the EC50 of nitrous oxide during three hours of exposure was 118 vol%. Vacuolization was induced by combining a nontoxic dose of ketamine (20 mg/kg) with nontoxic nitrous oxide (75 vol%) and the magnitude of the effect was similar to the maximum effect induced by ketamine alone at up to 80 mg/kg.
The ED50 of ketamine fell to 8.8 mg/kg when combined with nitrous oxide 120 vol% (Jevtovic-Todorovic et al., 2000). Likewise, ketamine (40 mg/kg) caused ineffective concentrations of nitrous oxide to become 16-fold (50 vol%) and 26-fold (75 vol%) more toxic. Ketamine (40 mg/kg) only produced 13 vacuolated neurons per section, but that increased to 288 when combined with nitrous oxide at 75 vol%. Toxicity was blocked by isoflurane (1.5 vol%), greatly reduced by halothane (1 vol%), and partially attenuated by diazepam (5 mg/kg IP).
Rats were susceptible to ketamine-induced neurotoxicity at all ages, but older animals were more vulnerable, whereas nitrous oxide had a similar effect across ages (Jevtovic-Todorovic and Carter, 2005). The ED50 of ketamine in young adult female rats was 66 mg/kg (IP), yet maximal toxicity had already occurred by 60 mg/kg in aging rats. Neither young nor aging animals were negatively affected by lower doses of ketamine (10-30 mg/kg). Ketamine and nitrous oxide only induced transient neurotoxic changes. PC/RSC neurons from young animals returned to normal 4 h after ketamine (60 mg/kg IP), although aging neurons, despite improving from the peak effect, still exhibited swelling and vacuolization at 5 h. This study also replicated the synergistic toxicity of ketamine and nitrous oxide reported by Jevtovic-Todorovic et al. (2000).
In the PC/RSC region of rats, acute and chronic (7-day) treatment with ketamine (50 mg/kg IP) or MK-801 (0.6 mg/kg IP) caused an increased in extracellular hydroxyl radicals; challenge with either drug following chronic exposure further increased hydroxyl radicals (Zuo et al., 2007). Both drugs caused hyperactivity with acute administration, a behavior that preceded the peak increase in hydroxyl radicals.
Subanesthetic ketamine (4-30 mg/kg IP) variabily affected indicators of oxidative stress throughout the brain in rats, as measured 30 min after administration (de Oliveira et al., 2009). At 4 mg/kg, lipid peroxidation increased in the cerebellum, hippocampus, PFC, striatum, and cerebral cortex; 10 mg/kg caused an increase in the cerebellum, striatum, and PFC. In contrast, 30 mg/kg was inactive on that measure in every region. Ketamine (10 mg/kg) increased carbonyl content (indicator of protein oxidation) in the hippocampus and striatum, but only in the hippocampus at 30 mg/kg, while causing a reduction in the PFC.
Sulfhydryl content was reduced in the hippocampus and cerebral cortex at 4 mg/kg; in the cerebellum, hippocampus, and striatum at 10 mg/kg; and in the cerebellum, striatum, and cerebral cortex at 30 mg/kg (de Oliveira et al., 2009). All doses reduced SOD activity in the PFC, cerebellum, hippocampus, and striatum, whereas there was an increase in the cerebral cortex at 30 mg/kg. Catalase activity was reduced at every dose in the cerebellum and PFC, as well as in the hippocampus at 10 mg/kg and in the cerebral cortex at 30 mg/kg, with no effect in the striatum.
Antidepressant doses of ketamine in mice increased lipid peroxidation (10 and 20 mg/kg IP), nitrite content (indicator of nitric oxide production) (20 mg/kg), and catalase activity (10 and 20 mg/kg), while reducing glutathione (5, 10, and 20 mg/kg) (da Silva et al., 2010). At these doses, FST and TST immobility were reduced, but an anxiogenic effect was observed in the EPM along with an increase in locomotion.
Heat shock protein 70 (HSP70), a marker of reversible neuronal damage, was increased by ketamine and S-ketamine in the retrosplenial cortex of rats, but R-ketamine was inactive at 25-75 mg/kg (IP) (Tian et al., 2018). At 24 h, S-ketamine did not increase HSP70 when given at 25 mg/kg, but it did at 50 and 75 mg/kg. Racemic ketamine increased HSP70 at 100 mg/kg. (+)-MK-801 (1 mg/kg) also increased HSP70.
PV-Expressing Neurons and Psychosis
NMDAR antagonist exposure has been associated with reduced prefrontal parvalbumin (PV) expression, a deficit reported in psychotic disorders (Beasley and Reynolds, 1997; Gonzalez-Burgos, 2015). The psychosis-like effects of ketamine, particularly during chronic use, could hypothetically be related in part to dysfunction of PV neurons.
Exposure to ketamine (0.5 μM) for 24 h increased neuronal superoxide production in PV interneurons and other cortical neurons, an effect blocked by the GABAA receptor agonist muscimol (Behrens et al., 2007). The effect was also attenuated by SOD-mimetic, supporting a role for superoxide, and the NADPH oxidase inhibitor apocynin reduced superoxide production and the loss of PV and GAD67 immunoreactivity in PV interneurons. Male mice given ketamine (30 mg/kg IP) on two consecutive days had increased cortical expression of the NADPH oxidase subunit Nox2 as well as the membrane protein p22phox when tested 18 h after the second dose. Superoxide was increased in the PFC, CA3, and thalamic reticular nucleus. The ketamine-induced increase in Nox2 and p22phox occurred alongside increased NADPH oxidase activity in cortical synaptosomes, whereas apocynin inhibited the increase.
R-ketamine (10 mg/kg IP) did not reduce PV immunoreactivity in the mPFC or DG of mice, whereas S-ketamine did, indicating the S-isomer is at least more potent for inducing this effect (Yang et al., 2015).
This was further demonstrated in mice with repeated administration of either isomer at 10 mg/kg (IP) eight times in a single week, with only S-ketamine reducing PV immunoreactivity (Yang et al., 2016). The reduction was observed one week after the last injection and was present in the mPFC, CA1, CA3, and DG; neither drug affected the NAc.
9.4 Abuse and Addiction
Opponents of ketamine and those who recommend a very cautious rollout frequently cite concerns about abuse and addiction because it has acutely rewarding effects and a small minority of users do become addicted. In the context of medical use, including for psychiatric conditions, the risk is small. However, there are at least a couple published reports in which ketamine use in a medical setting was subsequently associated with excessive use outside the confines of supervised dosing.
In one of the only published cases of ketamine abuse/addiction directly associated with depression treatment, the issues were largely attributable to poor management of the prescription since the physician provided ketamine for at-home use up to four times per day (Schak et al., 2016). That case does not demonstrate a problem with ketamine per se, though it illustrates why ketamine may not be a great medication outside hospital and clinics.
Bonnet (2015) reported a case of a female nurse with a long history of untreated MDD who received ketamine for pain relief when NSAIDs and tramadol were ineffective. Ketamine (25 mg IM) immediately reduced her pain and alleviated her depression for a week. Because she had not been given ketamine for depression, she began stealing it from her workplace when her symptoms returned, initially self-administering 50 mg (IM) per week. Those injections caused a brief 20-minute period of euphoria, relaxation, and psychedelic effects, followed by 5-8 days of depression relief. Within a couple months of weekly use, the acute effects faded and the duration of efficacy shortened to 1-2 days, leading to increased frequency and higher doses.
Her usage increased to 400 mg (IM) each day, which produced transient pleasant sensations but only one day of depression relief (Bonnet, 2015). She suppressed her depression for three months with this pattern of use; however, daily high-dose use caused cognitive impairment (e.g. reduced concentration, trouble finding words, impaired recall) and she tried to discontinue ketamine multiple times, in each instance returning to use within 2-3 days. She was admitted for treatment, at which time she did not have urological symptoms and only displayed poor concentration and slow thinking, but she left within a few days because of an increasing desire to use.
She was admitted a second time about a month later after increasing her use to 2 grams per day, which caused periods of unconsciousness, amnesia, and confusion (Bonnet, 2015). Her executive function, attention, and delayed recall were impaired; those issues resolved by Day 7 of admission. While she did not have depression at entrance (HDRS=7), that quickly changed, with her HDRS increasing to 27 during the first week. Because of severe depression and a loss of interest in treatment, she left. Finally, she was admitted a third time while on alcohol, which she had been using for depression while being unable to access ketamine. Her HDRS increased from 12 at admission to 32 by the third week of treatment. She agreed to pharmacotherapy with venlafaxine and remitted within three weeks without a continued urge to use ketamine; cognitive function was normal at that time.
In another case, an out-of-state physician prescribed ketamine to a male patient with a 30-year history of recurrent MDD (Schak et al., 2016). His baseline symptoms included suicidality and he had previously been hospitalized and treated with ECT. Months before one of his psychiatric admissions, he participated in a research study of IV ketamine, which produced 4-5 days of relief. He sought out a prescriber after leaving the study because of its efficacy; a neurologist agreed to prescribe him intranasal ketamine. It was shipped across state lines every month in the form of intranasal and sublingual formulations. Ketamine offered relief for a time, but it did not consistently address his depression and suicidality, and he still had to be hospitalized.
During one of his admissions, the patient admitted using more ketamine than prescribed (Schak et al., 2016). He had been told to use ketamine (75-150 mg intranasal) every four hours as needed, yet he was taking it 10-12 times per day because the benefits only lasted 2-3 hours; each dose caused dissociative and “trippy” effects for 20 minutes. His family was concerned about his use because he recklessly took ketamine while driving and it was “ripping his life apart.” Because he was increasing his use in a risky manner and had experienced loss of employment associated with it, the physicians treating him during his psychiatric admission considered him dependent on ketamine. After being contacted by the physicians, the prescriber agreed to end the prescription.
Since he believed ketamine was the only drug effective against his depression and he did not want to receive other treatments—including ECT because he experienced memory impairment after a prior instance of ECT therapy—he obtained ketamine elsewhere for a short time (Schak et al., 2016). A month later he was admitted after attempting suicide with alcohol and the hypnotic eszopiclone; he had not been using ketamine because it was inaccessible. Although the psychiatric team pushed for him to be involuntarily committed, the court did not agree. He died three months later in a car crash; ketamine was not found in his system, but alcohol, THC, and bupropion were. His family believed the cause of death was alcohol-facilitated suicide.
Grunebaum et al. (2018) did not find evidence of abuse during long-term follow-up of suicidal MDD patients who were treated with a single infusion of ketamine. Of 85 initial patients, 68 were reached at three months and 62 were reached at six months. Five (6%) reported receiving off-label ketamine at private clinics and one person had contemplated using ketamine provided by a friend; there were no instances of abuse.
Section 10: Comparison With Other Drugs
10.1 Glutamatergic Drugs
Riluzole
Riluzole is a medication used to slow the progression of amyotrophic lateral sclerosis (ALS). It is believed to work by reducing glutamate release, which can reduce excitotoxic damage in the CNS. Daily riluzole administration was researched as a way to extend ketamine-induced depression relief, but clinical studies have failed to detect a significant effect (Duncan et al., 2013; Mathew et al., 2010; Ibrahim et al., 2012).
Animal Research
Riluzole increased 13C labeling of glutamate, GABA, and glutamine in the PFC and hippocampus of male rats treated with 4 mg/kg/d IP for 21 days, as determined using MRS, indicating enhanced glutamate cycling (Chowdhury et al., 2008). This finding was unexpected because the mechanism of riluzole is thought to involve reduced glutamate release, although the increase may be consistent with greater glutamatergic metabolism, not reduced release.
Glial function was impaired in the PFC of male rats following CUS exposure, which occurred alongside behavioral deficits in the active avoidance test increased escape failures and sucrose preference (Banasr et al., 2010). They were exposed to CUS for 15 days and then received riluzole (4 mg/kg/d IP) for 21 days while stress continued. Glial deficits were shown by reduced acetate metabolism and reduced GFAP mRNA expression. Those effects were reversed by riluzole.
MK-801 (Dizocilpine)
Like ketamine, MK-801 is a noncompetitive NMDAR antagonist, but it is not approved for medical use. It primarily has acute antidepressant-like effects in animals, not extended ketamine-like effects. In this area of research, MK-801 usually refers to the more active (+)-enantiomer unless otherwise specified.
Animal Research
MK-801 had antidepressant effects in the mouse FST that were blocked by the AMPAR antagonist NBQX, but its duration of efficacy was relatively brief; ketamine (2.5 mg/kg IP) still had an effect two weeks after administration, whereas the effect of MK-801 was not sustained for a week (Maeng et al., 2008).
Similarly, a single dose of MK-801 was effective at reducing FST immobility in mice at 30 min and 3 h, but it was ineffective at 24 h (Autry et al., 2011). The NMDAR antagonist CPP remained effective at 24 h and ketamine (3 mg/kg IP) was effective for one week.
Immobility in the TST and FST in CSDS-susceptible mice was reduced by 0.1 mg/kg of (+)-MK-801 or (-)-MK-801, and sucrose preference was increased by (+)-MK-801 for 2-4 days after a single dose, with no effect by Day 7 (Yang et al., 2016).
Memantine (Namenda)
Memantine is an NMDAR antagonist with an affinity (Ki) of ~1.0 μM based on displacement of MK-801 in the human cortex, rat cortex, and rat hippocampal CA1 tissue (Parsons et al., 1999). It is approved for the treatment of Alzheimer’s disease. Memantine is largely free of psychotomimetic, dissociative, and hallucinatory effects at therapeutic doses. While it is an effective NMDAR antagonist when magnesium is not blocking the ion pore, i.e. during periods of greater neuronal activity, it is relatively ineffective near resting potentials and it does not inhibit currents induced by spontaneous glutamate release events, unlike ketamine (Gideons et al., 2014). Because NMDAR antagonism at rest can support synaptic plasticity, protein synthesis, AMPAR-mediated activity, and BDNF-TrkB signaling, memantine’s relative inefficacy under those conditions may be contributing to its overall inefficacy as an antidepressant.
Depression
Supporting Research
In a small 12-week, open-label study of flexible-dose memantine in MDD patients (n=8), depression was reduced, with a peak effect at Week 8 that was maintained through the rest of the trial, based on the seven study completers (Ferguson and Shingleton, 2007). The patients had moderate depression at baseline (MADRS = 32), but by the end of the study, the mean MADRS score was 19 (mild depression). Every patient reached at least 20 mg/d and three were titrated to 30 mg/d due to inefficacy by Week 8, two of whom were then increased to 40 mg/d after Week 10. Memantine was well-tolerated; the most common adverse effect was somnolence (n=3), followed by dizziness (n=2) and insomnia (n=2).
Kollmar et al. (2008) reported a significant reduction in depression and suicidality in a female patient treated with two ketamine infusions followed by daily memantine. She was hospitalized for a severe depressive episode involving at least 10 suicide attempts in the preceding two months; she was unresponsive to pharmacotherapy. ECT only offered partial relief and was accompanied by extended neuropsychological dysfunction. Ketamine (0.5 mg/kg IV, 40-min) substantially reduced her depression, but she was back to baseline by Day 4.
A second infusion two weeks later was similarly effective, but due to the ‘addictive’ properties of ketamine, the authors opted to prescribe memantine (Kollmar et al., 2008). Treatment was initiated with 5 mg/d, which was increased to 15 mg/d over the following four weeks. Her depression symptoms were greatly reduced during that period, producing a stable non-depressed HDRS of 7 and a BDI score of 5. She was discharged after 13 weeks and continued to receive memantine (15 mg/d) in addition to duloxetine, olanzapine, lorazepam, venlafaxine, mirtazapine, and lamotrigine; her condition remained stable six months later.
Opposing Research
Memantine failed to improve depression during an 8-week DBRCT of MDD patients treated with memantine (n=16) versus placebo (n=16); it was initiated at 5 mg/d and increased by 5 mg/week up to a max of 20 mg/d as tolerated (Zarate et al., 2006). The completion rate was 81% in both groups.
It was not superior to placebo in a 12-week DBRCT of patients over the age of 60 (n=35) who had depressive symptoms while in a rehab facility following a recent disabling medical event (e.g. orthopedic fracture) (Lenze et al., 2012). At baseline, patients had mild depression with an average HDRS of ~13; the completion rate was 77%. Depressive symptoms were similarly reduced in both groups without improvement in apathy. Functional recovery did not differ between groups.
Smith et al. (2013) found no effect of memantine augmentation in an 8-week DBRCT of MDD patients (n=31), 15 of whom were randomized to memantine; the completion rate was 84%, with one memantine patient discontinuing due to side effects and four placebo patients stopping because of side effects, increased depression, or change of location. Memantine was provided as a flexible dose of 5-20 mg/d and was added to existing antidepressant therapy. The mean MADRS change at Week 8 was -7.13 points from baseline in memantine-treated patients and -7.25 points in those who received placebo.
Alcohol Use Disorder
Opposing Research
The effects of memantine (20 mg/d) were compared with escitalopram during a 26-week DBRCT of 80 alcohol-dependent patients with comorbid MDD (Muhonen et al., 2008). The dropout rate was 28% (11/40) in each group. Both treatments were associated with reduced alcohol use and craving; no treatment had superior effects.
Traxoprodil (CP-101,606)
Traxoprodil is an NMDAR antagonist that is selective for receptors containing the GluN2B subunit. It was developed and researched by Pfizer, but it never became an approved medication.
Depression Research (Humans)
Supporting
Traxoprodil was more effective than placebo in a DBRCT of 30 TRD patients, producing a significant improvement on Day 4 (the main outcome) and most responders maintained their response for at least one week (Preskorn et al., 2008). Patients who did not respond to six weeks of paroxetine treatment were included; paroxetine was continued during the trial. A single infusion of Traxoprodil was used, specifically 0.5 mg/kg/h for 1.5 h. Initially it was given at 0.75 mg/kg/h for 1.5 h followed by 0.15 mg/kg/h for 6.5 h, but that caused moderate to severe dissociative effects in 4/7 subjects, so the dose was reduced for the remaining patients to improve blinding.
The Day 4 response rate was 60% compared with 20% in placebo-treated patients, with a mean MADRS score difference of 8.6 points (Preskorn et al., 2008). 78% of traxoprodil responders maintained their response for at least one week and 58% had a response on Day 11, 42% responded on Day 15, and 32% still had a response on Day 30. Patients who received the larger infusion dose seemed to experience the same antidepressant effect as those who were given the smaller dose. Six traxoprodil patients had dissociative reactions (2 mild, 2 moderate, 2 severe) compared with two mild reactions in placebo patients; the two patients with dissociation at the adjusted dose only had mild effects that were not substantially different from the dissociative-type effects in placebo patients.
Animal Research
Acute traxoprodil (5 or 15 mg/kg IV) dose-dependently produced a pattern of cortical and subcortical activation similar to what was observed with ketamine (3 mg/kg IV) in male rats (Tang et al., 2018). GluN2B-NMDAR occupancy was >80% in the forebrain with both traxoprodil doses at 5 and 30 min. At 5 mg/kg, activation was reported in the mPFC, ventral orbital cortex (VOC), and ACC, whereas the 15 mg/kg dose produced more widespread activation throughout cortical and subcortical regions, including the temporal cortex and striatum.
Lanicemine (AZD6765)
Lanicemine is a low-trapping NMDAR antagonist with an affinity similar to that of ketamine, displacing MK-801 with a Ki of 0.56-1.48 μM (Zarate et al., 2013). Its development was discontinued by AstraZeneca following failures in clinical research.
Depression Research (Human)
Supporting
A small, transient reduction in depression occurred after a single infusion of lanicemine (150 mg IV) compared to placebo in a DBRCT crossover study of 22 TRD patients (Zarate et al., 2013). MADRS score was improved within 80 min, but the benefit only remained significant through 110 min; it had a larger effect on HDRS ratings, where the difference was significant at 80 min, 110 min, and on Day 2. 32% of lanicemine patients responded during the trial compared with 15% of placebo patients. It did not cause psychotomimetic, manic, or dissociative effects. Plasma VEGF, but not BDNF, was increased after lanicemine.
Lanicemine (100 or 150 mg IV, three injections/week) was administered as an augmentation therapy in patients concurrently treated with up to two antidepressants who still had notable depressive symptoms (Sanacora et al., 2014). 51 MDD patients received 100 mg, 51 received 150 mg, and 50 were given placebo; treatment lasted for three weeks. Both doses were associated with a larger MADRS score reduction from baseline to Week 2, with efficacy detected after two weeks; the effects persisted for two weeks after the last infusion. Anxiety symptoms were also improved. Neither dose caused significant dissociative effects (CADSS) and dissociation-related effects were present in only 6-10% of lanicemine-treated patients. By Week 3, the response rates were: 16% with placebo, 37% with 100 mg lanicemine, and 29% with 150 mg.
Opposing
Lanicemine was ineffective in a phase 2 double-blind monotherapy study of 34 TRD patients treated with lanicemine (100 mg IV) (n=16) or placebo (n=18) (Sanacora et al., 2014). Lanicemine did not improve depression more than placebo at 24 h and there were only trends towards efficacy at 1 h and 72 h; however, there was a large placebo effect (MADRS change: -14.2 points) that could have made it more difficult to detect an effect. Lanicemine did not cause clinically meaningful dissociative effects, psychotomimetic effects, or cognitive impairment.
A larger DBRCT of 302 TRD patients who received lanicemine (50 or 100 mg) or placebo as an add-on to existing medication found no benefit following 15 infusions over a 12-week period (Sanacora et al., 2017). At Week 6, the response rates were: 39% for placebo, 36% for lanicemine 50 mg, and 44% for lanicemine 100 mg.
General Research (Humans)
A phase 1 DBRCT crossover study reported lanicemine’s effects in 23 healthy people; 14 received lanicemine 75 mg (IV), 19 received lanicemine 150 mg, 17 received ketamine, and 15 received placebo (Sanacora et al., 2014). Ketamine and lanicemine increased γ-band EEG activity and the effect of lanicemine 150 mg was effectively the same as ketamine; ketamine and lanicemine (150 mg) also reduced prefrontal theta-cordance, which could be a biomarker of antidepressant activity. While ketamine caused dissociation (CADSS), lanicemine did not.
The neurological effects of ketamine were distinct from lanicemine in a study of unmedicated MDD patients (n=56) randomized to placebo, ketamine (0.5 mg/kg IV), or lanicemine (100 mg IV) (Abdallah et al., 2018). Unlike ketamine, which increased PFC global brain connectivity with global signal regression (GBCr) during infusion and at 24 h, lanicemine had no effect at either timepoint.
Animal Research
Ketamine and lanicemine increased spontaneous γ-band activity in rats, as shown on EEG (Sanacora et al., 2013). Lanicemine (10 mg/kg IP) failed to produce antidepressant-like effects in CSDS-susceptible mice, whereas R-ketamine (10 mg/kg IP) was effective (Qu et al., 2017). R-ketamine also produced greater changes in the gut microbiome.
Rapastinel (GLYX-13)
Rapastinel is a tetrapeptide that was under development by Allergan. Some studies demonstrated antidepressant effects in humans and in animals, and its effects appeared to involve enhancement of NMDAR-mediated activity, increased synaptic plasticity, and rapid mTORC1 activation. In March 2019, Allergan announced that three Phase 3 clinical trials in MDD failed to meet their primary endpoint. Its development has been discontinued.
Depression Research (Humans)
Supporting
In a DBRCT of 116 MDD patients who had failed at least one standard antidepressant during their current episode, single-dose rapastinel was effective at 5 and 10 mg/kg (IV), but not at 1 or 30 mg/kg (Preskorn et al., 2015). Symptoms were reduced within two hours and remained lower through Days 1 and 7; there was also a large placebo response, with placebo patients showing a 45% reduction in HDRS-17 score at 24 h. Despite the large placebo effect, rapastinel was significantly effective at 5 and 10 mg/kg, while 1 mg/kg only caused a nonsignificant improvement. It failed to produce greater response or remission rates. Acute psychotomimetic and other significant side effects did not occur.
Animal Research
Rapastinel had antidepressant-like effects in rats that were dependent on AMPAR activity, with NBQX attenuating the benefits (Burgdorf et al., 2013). The SSRI fluoxetine only reduced floating time in the FST shortly after dosing, but rapastinel and ketamine remained effective 24 h and one week after injection. In the novelty-induced hypophagia (NIH) test, rapastinel and ketamine reduced latency to eat at 1 h. In vitro, both drugs increased LTP induced by high-frequency stimulation in hippocampal slices 24 h after exposure.
Rapastinel did not substitute for ketamine (10 mg/kg IV) in discrimination stimulus testing, it lacked rewarding effects in the CPP, there were no sensorimotor gating impairments in the PPI, and it did not cause sedation (Burgdorf et al., 2013). Both drugs increased surface levels of the GluN2B NMDAR subunit in the mPFC and hippocampus 24 h after dosing; they also increased surface levels of GluR1.
In reviewing the literature, Moskal and colleagues (2017) found positive effects of rapastinel on learning and memory in rats, as well as antidepressant effects in the FST, LH, and CUS models, which seemed to involve enhanced synaptic plasticity. There was an increase in mature dendritic spines in rat DG and layer V of the mPFC at 24 h, and ex vivo experiments demonstrated persistent effects on LTP two weeks after exposure.
The mTORC1 pathway was rapidly activated in the PFC of male rats following a single injection of rapastinel (3 mg/kg IV), as shown by increased p-mTOR, p-p70S6K1, p-4E-BP1, p-ERK, and p-Akt (Liu et al., 2017). Ketamine (10 mg/kg IP) caused similar effects. Rapastinel increased the number and function of spine synapses in the apical dendritic tuft of layer V pyramidal mPFC neurons and increased orexin-induced synaptic activity. Ketamine had properties that were potentially indicative of 5-HT2 receptor activity, namely increased impulsivity in the serial reaction time task (SRTT) and potentiation of head twitches induced by the 5-HT2 receptor agonist DOI at 24 h; rapastinel lacked those effects.
R-ketamine (10 mg/kg IP) and rapastinel (10 mg/kg IP) reduced immobility in the TST and FST, and increased sucrose preference in CSDS-susceptible mice up to one week after injection (Yang et al., 2016). Eight days after injection, R-ketamine attenuated a CSDS-induced deficit in BDNF-TrkB signaling, as well as PFC and hippocampal (DG and CA3) expression of PSD-95, GluR1; rapastinel was ineffective. Neither drug affected the increase in BDNF-TrkB signaling, PSD-95, and GluR1 in the NAc of susceptible mice. R-ketamine was more potent than rapastinel; 3 mg/kg of R-ketamine was effective on Day 7, but rapastinel was ineffective at that dose.
Rapastinel enhanced NMDAR activity and synaptic plasticity at antidepressant-relevant concentrations of 30-100 nM (Donello et al., 2019). Although it is frequently labeled a glycine site agonist, these effects came via an NMDAR domain that was distinct from the glycine site; D-serine did not affect rapastinel nor did glycine site antagonists block its activity as examined using rodent behavioral tests, microdialysis, calcium imaging, and electrophysiology (Donello et al., 2019). In vitro, 100 nM increased NMDAR-mediated EPSCs and enhanced LTP without affecting mEPSCs or paired-pulse facilitation, consistent with rapastinel working postsynaptically.
AGN-241751
Animal Research
In vitro testing demonstrated that AGN-241751 functions in a rapastinel-like way but with significantly higher potency (Banerjee et al., 2019). Testing was performed in magnesium-free medium with TTX and NBQX to isolate NMDAR activity. Calcium influx did not increase when AGN-241751 was applied on its own at 0.1-1000 nM, but when co-applied at 0.3-10 nM with NMDA, it potentiated NMDA-induced calcium influx by 30%; the effect did not appear to involve in the glycine site.
MK-801 binding was increased by 35 to 78% at AGN-241751 concentrations of 1 fM to 0.1 μM, with an EC50 ranging from 0.1 pM at GluN2C-NMDARs to 0.6 nM at GluN2B-NMDARs (Banerjee et al., 2019). LTP in mPFC neurons was increased at 20-100 nM. Oral bioavailability was very high (>95%) in rats and it produced a rapid dose-dependent reduction in FST immobility that persisted for 7-14 days; additionally, it increased mPFC plasticity.
Org-26576
Human Research
The AMPAR positive modulator Org-26576 appeared to enhance executive functioning and processing speed at 400 mg BID (oral) in a DBRCT of 30 MDD patients treated with 100 mg BID, 400 mg BID, or placebo (Nations et al., 2012). It was also studied in a separate group of patients (n=24) at 100-600 mg BID and was found to have a maximum tolerable dose of 450 mg BID. Both Org-26576 patient groups had nonsignificant depression changes versus placebo during the 28-day treatment period.
LY341495
LY341495, an mGluR2/3 antagonist, reduced FST immobility in male rats 1 and 24 h after exposure, which was associated with mTORC1 pathway activation and increased synaptic proteins, similar to ketamine (10 mg/kg IP) (Dwyer et al., 2012). Within one hour, there was an increase in p-mTOR, p-p70S6K, p-4E-BP1, and p-ERK in the PFC; the effect was smaller in the hippocampus, with the only significant change being an increase in p-ERK. Levels of PSD95, GluR1, and synapsin I were elevated in the PFC at 24 h, while there was a smaller increase in PSD95 and GluR1 in the hippocampus, without elevated synapsin I. Rapamycin prevented FST immobility reduction at 24 h, implicating the mTORC1 pathway.
Other
Clinical studies evaluated the effects of AV-101 (aka L-4-chlorokynurenine), a glycine site NMDAR antagonist that is not associated with ketamine-like side effects, but in May 2019, VistaGen announced that it failed to improve depression.
Research is ongoing for: NRX-101, a combination of D-cycloserine with the antipsychotic lurasidone; and AXS-05, a combination of the NMDAR antagonist DXM with the antidepressant bupropion (Hashimoto, 2019).
10.2 Scopolamine
Scopolamine is a nonselective muscarinic acetylcholine receptor (mAChR) antagonist currently used for motion sickness and postoperative nausea and vomiting (PONV); it is also known for its strong deliriant effects at high doses. It has rapid and persistent antidepressant-like effects in animals, has been efficacious in human trials, and appears to share multiple mechanisms with ketamine. Although its primary target in the CNS is different from that of ketamine, it appears to cause disinhibition through a mechanism involving GABAergic interneurons, like ketamine.
Supporting Human Research
In a DBRCT crossover study of 19 patients with recurrent MDD or BD, scopolamine (4 μg/kg IV, 15-min) was superior to placebo, producing a reduction in depression and anxiety (Furey and Drevets, 2006). Patients received three scopolamine or placebo sessions spaced 3-4 days from each other; 18/19 patients completed the trial. The scopolamine-first group exhibited a carryover of antidepressant efficacy when entering the placebo part of the study. Symptoms continued to improve with subsequent scopolamine infusions and all patients had at least a partial response at the end of the study, while 11/18 had a full response and 10/18 experienced remission. The side effects (scopolamine vs placebo) were: drowsiness (100% vs. 95%); dry mouth (100% vs. 63%); blurred vision (94% vs. 26%); lightheadedness (94% vs. 42%); dizziness (33% vs. 0%); hypotension (22% vs. 0%); diplopia (11% vs. 0%)
Drevets and Furey (2010) reported similar findings in a DBRCT crossover study of MDD patients (n=22) who received three scopolamine infusions (0.4 μg/kg IV, 15-min) compared with three placebo sessions spaced 3-5 days apart. Scopolamine reduced depressive symptoms (MADRS) by 32% in the first block, surpassing the 6.5% reduction caused by placebo. Carryover efficacy was observed in the second block, with scopolamine-first patients still exhibiting a reduction in depression before receiving placebo infusions. The scopolamine-second group had a 53% reduction in MADRS. 64% responded and 50% remitted by the end of the study. The primary acute side effects were drowsiness, blurred vision, dry mouth, lightheadedness, and reduced BP; no patient dropped out because of side effects.
Animal Research
A single dose of scopolamine (25 μg/kg IP) reduced FST immobility at 24 h, activated mTORC1 signaling, and increased the number and function of spine synapses in layer V PFC pyramidal neurons (Voleti et al., 2013). The antidepressant effects were blocked by rapamycin and NBQX. One hour after treatment, scopolamine caused a small increase in p-mTOR, p-Akt, and p-p70S6K, without affecting p-ERK; the increase in p-p70S6K was larger at 2 h. It increased the frequency and amplitude of serotonin- and orexin-induced EPSCs by 50-75% at 24 h. Both ketamine (10 mg/kg IP) and scopolamine transiently increased extracellular glutamate in the PFC within 10 min. Telenzepine, an antimuscarinic that is modestly selective for M1 muscarinic receptors over M3 receptors, had similar effects and was effective in the FST at 24 h.
Indirect activation of neuronal activity appeared to underly the effects of scopolamine, as knockdown of the M1 receptor on GABAergic interneurons in the mPFC, not glutamatergic neurons, attenuated the antidepressant-like effects of scopolamine (25 μg/kg IP; three injections, separated 48 h) in mice (Wohleb et al., 2016). Antidepressant-like behavioral effects were observed in the FST, NSFT, and FUST 24 h after the injection series. The M1 receptor was expressed on GABAergic (i.e. GAD67+) interneurons of the mPFC and on glutamatergic (CaMKII+) interneurons. In particular, SST interneurons rather than PV interneurons may have been involved; SST interneurons had greater M1 expression and knockdown of the receptor specifically on those interneurons interfered with the antidepressant-like behavioral effects.
Like with ketamine, increased BDNF signaling has also been implicated. The antidepressant-like effects of scopolamine (3 μg/kg IP; three doses, every other day) in mice were lost in BDNF Met/Met allele animals and when an anti-BDNF antibody was infused directly into the mPFC (Ghosal et al., 2018). Wild-type (WT) mice had an increase in TrkB phosphorylation in the PFC, but Met/Met mice did not, and pretreatment with the VDCC blocker verapamil attenuated scopolamine-induced antidepressant effects and BDNF-TrkB signaling.
Scopolamine rapidly increased BDNF release and TrkB signaling in primary cortical cultures, effects that were blocked by AMPAR antagonism with CNQX and by neuronal silencing with the GABAA receptor agonist muscimol (Ghosal et al., 2018). Incubation for 1 h at 1-100 nM increased p-ERK1/2, which was blocked by the TrkB inhibitor ANA-12 and by TTX. Similarly, scopolamine (3 nM) increased p-p70S6K and BDNF; TTX blocked those effects. The in vitro effects of scopolamine were replicated by the M1 selective antagonist VU0225035, which increased p-ERK and p-p70S6K, along with increasing BDNF release, which was blocked by TTX, CNQX, and muscimol. Incubation for 24 h at 1 nM increased dendritic complexity, with a greater number of dendrite branch crossings 50 and 100 microns from the soma.
10.3 GABAergic Drugs
If ketamine is largely functioning through an increase in activity mediated by disinhibition, its effects could be replicated by reducing GABAA receptor activity. Some research has focused on α5-containing GABAA receptors, which are prevalent in the PFC and hippocampus. The literature is mixed as to whether antidepressant effects are unique to GABAA receptor negative modulators and inverse agonists or if agonists/PAMs can be effective. Studies have supported both increased and decreased GABAA receptor function.
α5-GABAA Receptor Negative Modulators
Supporting Animal Research
Negative modulators of α5-GABAA receptors, namely L-655,708 and MRK-016, reversed chronic stress-induced deficits in social interaction and sucrose preference in rats (Fischell et al., 2015). L-655,708 also reversed a reduction in excitatory synaptic transmission and it increased GluR1 levels.
Consistent with that research, Zanos et al. (2017) observed FST immobility reduction 1 and 24 h after MRK-016, along with an anti-anhedonic effect in the FUST after chronic stress exposure. MRK-016, like ketamine, transiently increased EEG γ power; the EEG change and antidepressant effect were blocked by NBQX. It did not impair rotarod performance, cause a deficit in sensorimotor gating (PPI), alter locomotion, or induce rewarding effects (assessed using the CPP), unlike ketamine.
Allopregnanolone (Brexanolone; SAGE-547)
Allopregnanolone is an endogenous steroid hormone that is used as a medication for postpartum depression (PPD). It functions as a positive allosteric modulator (PAM) at synaptic and extrasynaptic GABAA receptors, particularly those with the δ subunit (Duman, 2018). Progesterone-derived neurosteroids like allopregnanolone are elevated during pregnancy and a large reduction in those hormones is thought to be a key mediator of PPD. Because PPD is a unique form of depression, it may respond to different treatments than typical MDD or BD.
Supporting Human Research
Administration of allopregnanolone to levels approximating those that occur during the third trimester of pregnancy caused remission in four women with severe PPD who were treated in an open-label manner; it was titrated for 12 h, maintained at a full dose for 36 h, and then titrated down for 12 h (Kanes et al., 2017).
A larger DBRCT of 21 inpatients with severe PPD found a continuous 60-hour IV infusion of allopregnanolone (n=10) was superior to placebo (n=11) at the end of the infusion (Kanes et al., 2017). Patients treated with allopregnanolone had a 21 point reduction in HDRS score, compared with 8 points in the placebo group.
10.4 mTOR
Navitor has developed an mTORC1 signaling enhancer called NV-5138 with the intent of using it in TRD. It increased mTORC1 signaling via activity at the upstream regulator sestrin and produced antidepressant-like effects in animals for up to one week in an mTORC1-dependent manner (Duman, 2018).
References
aan het Rot, M., Collins, K. A., Murrough, J. W., Perez, A. M., Reich, D. L., Charney, D. S., & Mathew, S. J. (2010). Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biological Psychiatry, 67(2), 139–145. https://doi.org/10.1016/j.biopsych.2009.08.038
Abdallah, C. G., Averill, L. A., Gueorguieva, R., Goktas, S., Purohit, P., Ranganathan, M., Sherif, M., Ahn, K.-H., D’Souza, D. C., Formica, R., Southwick, S. M., Duman, R. S., Sanacora, G., & Krystal, J. H. (2020). Modulation of the antidepressant effects of ketamine by the mTORC1 inhibitor rapamycin. Neuropsychopharmacology, 45(6), 990–997. https://doi.org/10.1038/s41386-020-0644-9
Abdallah, C. G., Dutta, A., Averill, C. L., McKie, S., Akiki, T. J., Averill, L. A., & Deakin, J. F. W. (2018). Ketamine, but Not the NMDAR Antagonist Lanicemine, Increases Prefrontal Global Connectivity in Depressed Patients. Chronic Stress (Thousand Oaks, Calif.), 2. https://doi.org/10.1177/2470547018796102
Abdallah, C. G., Fasula, M., Kelmendi, B., Sanacora, G., & Ostroff, R. (2012). Rapid antidepressant effect of ketamine in the electroconvulsive therapy setting. The Journal of ECT, 28(3), 157–161. https://doi.org/10.1097/YCT.0b013e31824f8296
Abdallah, C. G., Jackowski, A., Salas, R., Gupta, S., Sato, J. R., Mao, X., Coplan, J. D., Shungu, D. C., & Mathew, S. J. (2017). The Nucleus Accumbens and Ketamine Treatment in Major Depressive Disorder. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 42(8), 1739–1746. https://doi.org/10.1038/npp.2017.49
Abdallah, C. G., Salas, R., Jackowski, A., Baldwin, P., Sato, J. R., & Mathew, S. J. (2015). Hippocampal volume and the rapid antidepressant effect of ketamine. Journal of Psychopharmacology (Oxford, England), 29(5), 591–595. https://doi.org/10.1177/0269881114544776
Ajub, E., & Lacerda, A. L. T. (2018). Efficacy of Esketamine in the Treatment of Depression With Psychotic Features: A Case Series. Biological Psychiatry, 83(1), e15–e16. https://doi.org/10.1016/j.biopsych.2017.06.011
Akinfiresoye, L., & Tizabi, Y. (2013). Antidepressant effects of AMPA and ketamine combination: Role of hippocampal BDNF, synapsin, and mTOR. Psychopharmacology, 230(2), 291–298. https://doi.org/10.1007/s00213-013-3153-2
Albott, C. S., Lim, K. O., Forbes, M. K., Erbes, C., Tye, S. J., Grabowski, J. G., Thuras, P., Batres-Y-Carr, T. M., Wels, J., & Shiroma, P. R. (2018). Efficacy, Safety, and Durability of Repeated Ketamine Infusions for Comorbid Posttraumatic Stress Disorder and Treatment-Resistant Depression. The Journal of Clinical Psychiatry, 79(3). https://doi.org/10.4088/JCP.17m11634
Allen, A. P., Naughton, M., Dowling, J., Walsh, A., Ismail, F., Shorten, G., Scott, L., McLoughlin, D. M., Cryan, J. F., Dinan, T. G., & Clarke, G. (2015). Serum BDNF as a peripheral biomarker of treatment-resistant depression and the rapid antidepressant response: A comparison of ketamine and ECT. Journal of Affective Disorders, 186, 306–311. https://doi.org/10.1016/j.jad.2015.06.033
Arabzadeh, S., Hakkikazazi, E., Shahmansouri, N., Tafakhori, A., Ghajar, A., Jafarinia, M., & Akhondzadeh, S. (2018). Does oral administration of ketamine accelerate response to treatment in major depressive disorder? Results of a double-blind controlled trial. Journal of Affective Disorders, 235, 236–241. https://doi.org/10.1016/j.jad.2018.02.056
Autry, A. E., Adachi, M., Nosyreva, E., Na, E. S., Los, M. F., Cheng, P., Kavalali, E. T., & Monteggia, L. M. (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature, 475(7354), 91–95. https://doi.org/10.1038/nature10130
Autry, A. E., & Monteggia, L. M. (2012). Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacological Reviews, 64(2), 238–258. https://doi.org/10.1124/pr.111.005108
Baldessarini, R. J., Tondo, L., Davis, P., Pompili, M., Goodwin, F. K., & Hennen, J. (2006). Decreased risk of suicides and attempts during long-term lithium treatment: A meta-analytic review. Bipolar Disorders, 8(5 Pt 2), 625–639. https://doi.org/10.1111/j.1399-5618.2006.00344.x
Ballard, E. D., Ionescu, D. F., Vande Voort, J. L., Niciu, M. J., Richards, E. M., Luckenbaugh, D. A., Brutsché, N. E., Ameli, R., Furey, M. L., & Zarate, C. A. (2014). Improvement in suicidal ideation after ketamine infusion: Relationship to reductions in depression and anxiety. Journal of Psychiatric Research, 58, 161–166. https://doi.org/10.1016/j.jpsychires.2014.07.027
Ban, T. A. (2006). The role of serendipity in drug discovery. Clinical Research, 8(3), 10.
Banasr, M., Chowdhury, G. M. I., Terwilliger, R., Newton, S. S., Duman, R. S., Behar, K. L., & Sanacora, G. (2010). Glial pathology in an animal model of depression: Reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Molecular Psychiatry, 15(5), 501–511. https://doi.org/10.1038/mp.2008.106
Banerjee, P., Donello, J., Li, Y.-X., Bertelsen, K., Guo, Y.-X., Zhang, X.-L., Stanton, P., Kroes, R., Gross, A., & Moskal, J. (2019). S133. AGN-241751, an Orally Bioavailable Positive NMDA Receptor Modulator, Exhibits Rapid and Sustained Antidepressant-Like Effects in Rodents. Biological Psychiatry, 85(10), S348. https://doi.org/10.1016/j.biopsych.2019.03.884
Bansinath, M., Warner, W., Tang, C. K., Turndorf, H., & Puig, M. M. (1992). On the mechanism of the interaction of ketamine and halothane in vitro. General Pharmacology, 23(6), 1183–1187. https://doi.org/10.1016/0306-3623(92)90309-8
Bartoli, F., Riboldi, I., Crocamo, C., Di Brita, C., Clerici, M., & Carrà, G. (2017). Ketamine as a rapid-acting agent for suicidal ideation: A meta-analysis. Neuroscience and Biobehavioral Reviews, 77, 232–236. https://doi.org/10.1016/j.neubiorev.2017.03.010
Baune, B. T., Miller, R., McAfoose, J., Johnson, M., Quirk, F., & Mitchell, D. (2010). The role of cognitive impairment in general functioning in major depression. Psychiatry Research, 176(2–3), 183–189. https://doi.org/10.1016/j.psychres.2008.12.001
Beasley, C., & Reynolds, G. (1997). Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophrenia Research, 24(3), 349–355. https://doi.org/10.1016/S0920-9964(96)00122-3
Beck, A. T., & Beamesderfer, A. (1974). Assessment of depression: The depression inventory. Modern Problems of Pharmacopsychiatry, 7(0), 151–169. https://doi.org/10.1159/000395074
Beck, A. T., Ward, C. H., Mendelson, M., Mock, J., & Erbaugh, J. (1961). An inventory for measuring depression. Archives of General Psychiatry, 4, 561–571. https://doi.org/10.1001/archpsyc.1961.01710120031004
Beck, Aaron T., Kovacs, M., & Weissman, A. (1979). Assessment of suicidal intention: The Scale for Suicide Ideation. Journal of Consulting and Clinical Psychology, 47(2), 343–352. https://doi.org/10.1037/0022-006X.47.2.343
Beck, Aaron T., Steer, R. A., & Carbin, M. G. (1988). Psychometric properties of the Beck Depression Inventory: Twenty-five years of evaluation. Clinical Psychology Review, 8(1), 77–100. https://doi.org/10.1016/0272-7358(88)90050-5
Behrens, M. M., Ali, S. S., Dao, D. N., Lucero, J., Shekhtman, G., Quick, K. L., & Dugan, L. L. (2007). Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science (New York, N.Y.), 318(5856), 1645–1647. https://doi.org/10.1126/science.1148045
Belujon, P., & Grace, A. A. (2014). Restoring mood balance in depression: Ketamine reverses deficit in dopamine-dependent synaptic plasticity. Biological Psychiatry, 76(12), 927–936. https://doi.org/10.1016/j.biopsych.2014.04.014
Beneyto, M., Kristiansen, L. V., Oni-Orisan, A., McCullumsmith, R. E., & Meador-Woodruff, J. H. (2007). Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 32(9), 1888–1902. https://doi.org/10.1038/sj.npp.1301312
Berman, R. M., Cappiello, A., Anand, A., Oren, D. A., Heninger, G. R., Charney, D. S., & Krystal, J. H. (2000). Antidepressant effects of ketamine in depressed patients. Biological Psychiatry, 47(4), 351–354. https://doi.org/10.1016/s0006-3223(99)00230-9
Berton, O., McClung, C. A., Dileone, R. J., Krishnan, V., Renthal, W., Russo, S. J., Graham, D., Tsankova, N. M., Bolanos, C. A., Rios, M., Monteggia, L. M., Self, D. W., & Nestler, E. J. (2006). Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science (New York, N.Y.), 311(5762), 864–868. https://doi.org/10.1126/science.1120972
Bessa, J. M., Ferreira, D., Melo, I., Marques, F., Cerqueira, J. J., Palha, J. A., Almeida, O. F. X., & Sousa, N. (2009). The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Molecular Psychiatry, 14(8), 764–773. https://doi.org/10.1038/mp.2008.119
Beurel, E., Song, L., & Jope, R. S. (2011). Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Molecular Psychiatry, 16(11), 1068–1070. https://doi.org/10.1038/mp.2011.47
Beurel, Eleonore, Grieco, S. F., & Jope, R. S. (2015). Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacology & Therapeutics, 148, 114–131. https://doi.org/10.1016/j.pharmthera.2014.11.016
Björkholm, C., Jardemark, K., Schilström, B., & Svensson, T. H. (2015). Ketamine-like effects of a combination of olanzapine and fluoxetine on AMPA and NMDA receptor-mediated transmission in the medial prefrontal cortex of the rat. European Neuropsychopharmacology: The Journal of the European College of Neuropsychopharmacology, 25(10), 1842–1847. https://doi.org/10.1016/j.euroneuro.2015.07.002
Blake, D. D., Weathers, F. W., Nagy, L. M., Kaloupek, D. G., Gusman, F. D., Charney, D. S., & Keane, T. M. (1995). The development of a Clinician-Administered PTSD Scale. Journal of Traumatic Stress, 8(1), 75–90. https://doi.org/10.1007/BF02105408
Blasco-Serra, A., González-Soler, E. M., Cervera-Ferri, A., Teruel-Martí, V., & Valverde-Navarro, A. A. (2017). A standardization of the Novelty-Suppressed Feeding Test protocol in rats. Neuroscience Letters, 658, 73–78. https://doi.org/10.1016/j.neulet.2017.08.019
Blier, P., Zigman, D., & Blier, J. (2012). On the safety and benefits of repeated intravenous injections of ketamine for depression. Biological Psychiatry, 72(4), e11-12. https://doi.org/10.1016/j.biopsych.2012.02.039
Bloch, M. H., Wasylink, S., Landeros-Weisenberger, A., Panza, K. E., Billingslea, E., Leckman, J. F., Krystal, J. H., Bhagwagar, Z., Sanacora, G., & Pittenger, C. (2012). Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biological Psychiatry, 72(11), 964–970. https://doi.org/10.1016/j.biopsych.2012.05.028
Bonhomme, V., Vanhaudenhuyse, A., Demertzi, A., Bruno, M.-A., Jaquet, O., Bahri, M. A., Plenevaux, A., Boly, M., Boveroux, P., Soddu, A., Brichant, J. F., Maquet, P., & Laureys, S. (2016). Resting-state Network-specific Breakdown of Functional Connectivity during Ketamine Alteration of Consciousness in Volunteers. Anesthesiology, 125(5), 873–888. https://doi.org/10.1097/ALN.0000000000001275
Bonnet, U. (2015). Long-Term Ketamine Self-Injections in Major Depressive Disorder: Focus on Tolerance in Ketamine’s Antidepressant Response and the Development of Ketamine Addiction. Journal of Psychoactive Drugs, 47(4), 276–285. https://doi.org/10.1080/02791072.2015.1072653
Bora, E., Harrison, B. J., Yücel, M., & Pantelis, C. (2013). Cognitive impairment in euthymic major depressive disorder: A meta-analysis. Psychological Medicine, 43(10), 2017–2026. https://doi.org/10.1017/S0033291712002085
Breier, A., Malhotra, A. K., Pinals, D. A., Weisenfeld, N. I., & Pickar, D. (1997). Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. The American Journal of Psychiatry, 154(6), 805–811. https://doi.org/10.1176/ajp.154.6.805
Bremner, J. D., Krystal, J. H., Putnam, F. W., Southwick, S. M., Marmar, C., Charney, D. S., & Mazure, C. M. (1998). Measurement of dissociative states with the Clinician-Administered Dissociative States Scale (CADSS). Journal of Traumatic Stress, 11(1), 125–136. https://doi.org/10.1023/A:1024465317902
Brinck, E., Tiippana, E., Heesen, M., Bell, R. F., Straube, S., Moore, R. A., & Kontinen, V. (2018). Perioperative intravenous ketamine for acute postoperative pain in adults. Cochrane Database of Systematic Reviews. https://doi.org/10.1002/14651858.CD012033.pub4
Burgdorf, J., Zhang, X., Nicholson, K. L., Balster, R. L., Leander, J. D., Stanton, P. K., Gross, A. L., Kroes, R. A., & Moskal, J. R. (2013). GLYX-13, a NMDA receptor glycine-site functional partial agonist, induces antidepressant-like effects without ketamine-like side effects. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 38(5), 729–742. https://doi.org/10.1038/npp.2012.246
Busk, J., Faurholt-Jepsen, M., Frost, M., Bardram, J. E., Kessing, L. V., & Winther, O. (2020). Daily estimates of clinical severity of symptoms in bipolar disorder from smartphone-based self-assessments. Translational Psychiatry, 10(1), 194. https://doi.org/10.1038/s41398-020-00867-6
Cabib, S., & Puglisi-Allegra, S. (2012). The mesoaccumbens dopamine in coping with stress. Neuroscience and Biobehavioral Reviews, 36(1), 79–89. https://doi.org/10.1016/j.neubiorev.2011.04.012
Can, A., Dao, D. T., Terrillion, C. E., Piantadosi, S. C., Bhat, S., & Gould, T. D. (2012). The tail suspension test. Journal of Visualized Experiments: JoVE, 59, e3769. https://doi.org/10.3791/3769
Can, A., Zanos, P., Moaddel, R., Kang, H. J., Dossou, K. S. S., Wainer, I. W., Cheer, J. F., Frost, D. O., Huang, X.-P., & Gould, T. D. (2016). Effects of Ketamine and Ketamine Metabolites on Evoked Striatal Dopamine Release, Dopamine Receptors, and Monoamine Transporters. The Journal of Pharmacology and Experimental Therapeutics, 359(1), 159–170. https://doi.org/10.1124/jpet.116.235838
Carlson, P. J., Diazgranados, N., Nugent, A. C., Ibrahim, L., Luckenbaugh, D. A., Brutsche, N., Herscovitch, P., Manji, H. K., Zarate, C. A., & Drevets, W. C. (2013). Neural correlates of rapid antidepressant response to ketamine in treatment-resistant unipolar depression: A preliminary positron emission tomography study. Biological Psychiatry, 73(12), 1213–1221. https://doi.org/10.1016/j.biopsych.2013.02.008
Carreno, F. R., Donegan, J. J., Boley, A. M., Shah, A., DeGuzman, M., Frazer, A., & Lodge, D. J. (2016). Activation of a ventral hippocampus-medial prefrontal cortex pathway is both necessary and sufficient for an antidepressant response to ketamine. Molecular Psychiatry, 21(9), 1298–1308. https://doi.org/10.1038/mp.2015.176
Carrier, N., & Kabbaj, M. (2013). Sex differences in the antidepressant-like effects of ketamine. Neuropharmacology, 70, 27–34. https://doi.org/10.1016/j.neuropharm.2012.12.009
Cavalleri, L., Merlo Pich, E., Millan, M. J., Chiamulera, C., Kunath, T., Spano, P. F., & Collo, G. (2018). Ketamine enhances structural plasticity in mouse mesencephalic and human iPSC-derived dopaminergic neurons via AMPAR-driven BDNF and mTOR signaling. Molecular Psychiatry, 23(4), 812–823. https://doi.org/10.1038/mp.2017.241
Chen, X., Shu, S., & Bayliss, D. A. (2009). HCN1 channel subunits are a molecular substrate for hypnotic actions of ketamine. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 29(3), 600–609. https://doi.org/10.1523/JNEUROSCI.3481-08.2009
Chilukuri, H., Reddy, N. P., Pathapati, R. M., Manu, A. N., Jollu, S., & Shaik, A. B. (2014). Acute antidepressant effects of intramuscular versus intravenous ketamine. Indian Journal of Psychological Medicine, 36(1), 71–76. https://doi.org/10.4103/0253-7176.127258
Chiu, C.-T., Scheuing, L., Liu, G., Liao, H.-M., Linares, G. R., Lin, D., & Chuang, D.-M. (2014). The mood stabilizer lithium potentiates the antidepressant-like effects and ameliorates oxidative stress induced by acute ketamine in a mouse model of stress. The International Journal of Neuropsychopharmacology, 18(6). https://doi.org/10.1093/ijnp/pyu102
Cho, J. E., Shim, J. K., Choi, Y. S., Kim, D. H., Hong, S. W., & Kwak, Y. L. (2009). Effect of low-dose ketamine on inflammatory response in off-pump coronary artery bypass graft surgery. British Journal of Anaesthesia, 102(1), 23–28. https://doi.org/10.1093/bja/aen325
Choi, M., Lee, S. H., Wang, S. E., Ko, S. Y., Song, M., Choi, J.-S., Kim, Y.-S., Duman, R. S., & Son, H. (2015). Ketamine produces antidepressant-like effects through phosphorylation-dependent nuclear export of histone deacetylase 5 (HDAC5) in rats. Proceedings of the National Academy of Sciences, 112(51), 15755–15760. https://doi.org/10.1073/pnas.1513913112
Chong, C., Schug, S. A., Page-Sharp, M., Jenkins, B., & Ilett, K. F. (2009). Development of a Sublingual/Oral Formulation of Ketamine for Use in Neuropathic Pain: Preliminary Findings from a Three-Way Randomized, Crossover Study. Clinical Drug Investigation, 29(5), 317–324. https://doi.org/10.2165/00044011-200929050-00004
Chou, D., Peng, H.-Y., Lin, T.-B., Lai, C.-Y., Hsieh, M.-C., Wen, Y.-C., Lee, A.-S., Wang, H.-H., Yang, P.-S., Chen, G.-D., & Ho, Y.-C. (2018). (2R,6R)-hydroxynorketamine rescues chronic stress-induced depression-like behavior through its actions in the midbrain periaqueductal gray. Neuropharmacology, 139, 1–12. https://doi.org/10.1016/j.neuropharm.2018.06.033
Chowdhury, G. M. I., Zhang, J., Thomas, M., Banasr, M., Ma, X., Pittman, B., Bristow, L., Schaeffer, E., Duman, R. S., Rothman, D. L., Behar, K. L., & Sanacora, G. (2017). Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Molecular Psychiatry, 22(1), 120–126. https://doi.org/10.1038/mp.2016.34
Chowdhury, Golam M. I., Banasr, M., de Graaf, R. A., Rothman, D. L., Behar, K. L., & Sanacora, G. (2008). Chronic riluzole treatment increases glucose metabolism in rat prefrontal cortex and hippocampus. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism, 28(12), 1892–1897. https://doi.org/10.1038/jcbfm.2008.78
Chowdhury, Golam M. I., Behar, K. L., Cho, W., Thomas, M. A., Rothman, D. L., & Sanacora, G. (2012). 1H-[13C]-nuclear magnetic resonance spectroscopy measures of ketamine’s effect on amino acid neurotransmitter metabolism. Biological Psychiatry, 71(11), 1022–1025. https://doi.org/10.1016/j.biopsych.2011.11.006
Chu, P. S.-K., Ma, W.-K., Wong, S. C.-W., Chu, R. W.-H., Cheng, C.-H., Wong, S., Tse, J. M.-L., Lau, F.-L., Yiu, M.-K., & Man, C.-W. (2008). The destruction of the lower urinary tract by ketamine abuse: A new syndrome? BJU International, 102(11), 1616–1622. https://doi.org/10.1111/j.1464-410X.2008.07920.x
Cipriani, A., Pretty, H., Hawton, K., & Geddes, J. R. (2005). Lithium in the prevention of suicidal behavior and all-cause mortality in patients with mood disorders: A systematic review of randomized trials. The American Journal of Psychiatry, 162(10), 1805–1819. https://doi.org/10.1176/appi.ajp.162.10.1805
Clark-Raymond, A., & Halaris, A. (2013). VEGF and depression: A comprehensive assessment of clinical data. Journal of Psychiatric Research, 47(8), 1080–1087. https://doi.org/10.1016/j.jpsychires.2013.04.008
Clements, J. A., Nimmo, W. S., & Grant, I. S. (1982). Bioavailability, Pharmacokinetics, and Analgesic Activity of Ketamine in Humans. Journal of Pharmaceutical Sciences, 71(5), 539–542. https://doi.org/10.1002/jps.2600710516
Cohen, M. L., Chan, S. L., Way, W. L., & Trevor, A. J. (1973). Distribution in the brain and metabolism of ketamine in the rat after intravenous administration. Anesthesiology, 39(4), 370–376. https://doi.org/10.1097/00000542-197310000-00003
Collo, G., Cavalleri, L., Chiamulera, C., & Merlo Pich, E. (2018). (2R,6R)-Hydroxynorketamine promotes dendrite outgrowth in human inducible pluripotent stem cell-derived neurons through AMPA receptor with timing and exposure compatible with ketamine infusion pharmacokinetics in humans. Neuroreport, 29(16), 1425–1430. https://doi.org/10.1097/WNR.0000000000001131
Correia-Melo, F. S., Argolo, F. C., Araújo-de-Freitas, L., Leal, G. C., Kapczinski, F., Lacerda, A. L., & Quarantini, L. C. (2017). Rapid infusion of esketamine for unipolar and bipolar depression: A retrospective chart review. Neuropsychiatric Disease and Treatment, 13, 1627–1632. https://doi.org/10.2147/NDT.S135623
Cotter, D., Mackay, D., Landau, S., Kerwin, R., & Everall, I. (2001). Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Archives of General Psychiatry, 58(6), 545–553. https://doi.org/10.1001/archpsyc.58.6.545
Cotter, David, Mackay, D., Chana, G., Beasley, C., Landau, S., & Everall, I. P. (2002). Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cerebral Cortex (New York, N.Y.: 1991), 12(4), 386–394. https://doi.org/10.1093/cercor/12.4.386
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., & Hemmings, B. A. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature, 378(6559), 785–789. https://doi.org/10.1038/378785a0
Cui, Y., Yang, Y., Ni, Z., Dong, Y., Cai, G., Foncelle, A., Ma, S., Sang, K., Tang, S., Li, Y., Shen, Y., Berry, H., Wu, S., & Hu, H. (2018). Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature, 554(7692), 323–327. https://doi.org/10.1038/nature25752
Curtin, S. C., Warner, M., & Hedegaard, H. (2016). Increase in Suicide in the United States, 1999-2014. NCHS Data Brief, 241, 1–8.
Cusin, C., Hilton, G. Q., Nierenberg, A. A., & Fava, M. (2012). Long-term maintenance with intramuscular ketamine for treatment-resistant bipolar II depression. The American Journal of Psychiatry, 169(8), 868–869. https://doi.org/10.1176/appi.ajp.2012.12020219
da Frota Ribeiro, C. M., Sanacora, G., Hoffman, R., & Ostroff, R. (2016). The Use of Ketamine for the Treatment of Depression in the Context of Psychotic Symptoms: To the Editor. Biological Psychiatry, 79(9), e65–e66. https://doi.org/10.1016/j.biopsych.2015.05.016
da Silva, F. C. C., do Carmo de Oliveira Cito, M., da Silva, M. I. G., Moura, B. A., de Aquino Neto, M. R., Feitosa, M. L., de Castro Chaves, R., Macedo, D. S., de Vasconcelos, S. M. M., de França Fonteles, M. M., & de Sousa, F. C. F. (2010). Behavioral alterations and pro-oxidant effect of a single ketamine administration to mice. Brain Research Bulletin, 83(1–2), 9–15. https://doi.org/10.1016/j.brainresbull.2010.05.011
Dakwar, E., Anerella, C., Hart, C. L., Levin, F. R., Mathew, S. J., & Nunes, E. V. (2014). Therapeutic infusions of ketamine: Do the psychoactive effects matter? Drug and Alcohol Dependence, 136, 153–157. https://doi.org/10.1016/j.drugalcdep.2013.12.019
Dakwar, Elias, Levin, F., Foltin, R. W., Nunes, E. V., & Hart, C. L. (2014). The effects of subanesthetic ketamine infusions on motivation to quit and cue-induced craving in cocaine-dependent research volunteers. Biological Psychiatry, 76(1), 40–46. https://doi.org/10.1016/j.biopsych.2013.08.009
D’Andrea, D., & Andrew Sewell, R. (2013). Transient resolution of treatment-resistant posttraumatic stress disorder following ketamine infusion. Biological Psychiatry, 74(9), e13-14. https://doi.org/10.1016/j.biopsych.2013.04.019
Davies, S. N., & Lodge, D. (1987). Evidence for involvement of N-methylaspartate receptors in “wind-up” of class 2 neurones in the dorsal horn of the rat. Brain Research, 424(2), 402–406. https://doi.org/10.1016/0006-8993(87)91487-9
de Beurs, D. P., Fokkema, M., & O’Connor, R. C. (2016). Optimizing the assessment of suicidal behavior: The application of curtailment techniques. Journal of Affective Disorders, 196, 218–224. https://doi.org/10.1016/j.jad.2016.02.033
de Oliveira, L., Spiazzi, C. M. dos S., Bortolin, T., Canever, L., Petronilho, F., Mina, F. G., Dal-Pizzol, F., Quevedo, J., & Zugno, A. I. (2009). Different sub-anesthetic doses of ketamine increase oxidative stress in the brain of rats. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 33(6), 1003–1008. https://doi.org/10.1016/j.pnpbp.2009.05.010
Deakin, J. F. W., Lees, J., McKie, S., Hallak, J. E. C., Williams, S. R., & Dursun, S. M. (2008). Glutamate and the neural basis of the subjective effects of ketamine: A pharmaco-magnetic resonance imaging study. Archives of General Psychiatry, 65(2), 154–164. https://doi.org/10.1001/archgenpsychiatry.2007.37
DeLorenzo, C., DellaGioia, N., Bloch, M., Sanacora, G., Nabulsi, N., Abdallah, C., Yang, J., Wen, R., Mann, J. J., Krystal, J. H., Parsey, R. V., Carson, R. E., & Esterlis, I. (2015). In vivo ketamine-induced changes in [11C]ABP688 binding to metabotropic glutamate receptor subtype 5. Biological Psychiatry, 77(3), 266–275. https://doi.org/10.1016/j.biopsych.2014.06.024
Denk, M. C., Rewerts, C., Holsboer, F., Erhardt-Lehmann, A., & Turck, C. W. (2011). Monitoring ketamine treatment response in a depressed patient via peripheral mammalian target of rapamycin activation. The American Journal of Psychiatry, 168(7), 751–752. https://doi.org/10.1176/appi.ajp.2011.11010128
Deyama, S., Bang, E., Kato, T., Li, X.-Y., & Duman, R. S. (2019). Neurotrophic and Antidepressant Actions of Brain-Derived Neurotrophic Factor Require Vascular Endothelial Growth Factor. Biological Psychiatry, 86(2), 143–152. https://doi.org/10.1016/j.biopsych.2018.12.014
Deyama, S., Bang, E., Wohleb, E. S., Li, X.-Y., Kato, T., Gerhard, D. M., Dutheil, S., Dwyer, J. M., Taylor, S. R., Picciotto, M. R., & Duman, R. S. (2019). Role of Neuronal VEGF Signaling in the Prefrontal Cortex in the Rapid Antidepressant Effects of Ketamine. The American Journal of Psychiatry, 176(5), 388–400. https://doi.org/10.1176/appi.ajp.2018.17121368
Diamond, P. R., Farmery, A. D., Atkinson, S., Haldar, J., Williams, N., Cowen, P. J., Geddes, J. R., & McShane, R. (2014). Ketamine infusions for treatment resistant depression: A series of 28 patients treated weekly or twice weekly in an ECT clinic. Journal of Psychopharmacology (Oxford, England), 28(6), 536–544. https://doi.org/10.1177/0269881114527361
DiazGranados, N., Ibrahim, L. A., Brutsche, N. E., Ameli, R., Henter, I. D., Luckenbaugh, D. A., Machado-Vieira, R., & Zarate, C. A. (2010). Rapid resolution of suicidal ideation after a single infusion of an N-methyl-D-aspartate antagonist in patients with treatment-resistant major depressive disorder. The Journal of Clinical Psychiatry, 71(12), 1605–1611. https://doi.org/10.4088/JCP.09m05327blu
Diazgranados, N., Ibrahim, L., Brutsche, N. E., Newberg, A., Kronstein, P., Khalife, S., Kammerer, W. A., Quezado, Z., Luckenbaugh, D. A., Salvadore, G., Machado-Vieira, R., Manji, H. K., & Zarate, C. A. (2010). A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Archives of General Psychiatry, 67(8), 793–802. https://doi.org/10.1001/archgenpsychiatry.2010.90
Domino, E. F., Chodoff, P., & Corssen, G. (1965). Pharmacologic effects of CI-581, a new dissociative anesthetic, in man. Clinical Pharmacology and Therapeutics, 6, 279–291. https://doi.org/10.1002/cpt196563279
Domino, Edward F. (2010). Taming the Ketamine Tiger: Anesthesiology, 1. https://doi.org/10.1097/ALN.0b013e3181ed09a2
Donello, J. E., Banerjee, P., Li, Y.-X., Guo, Y.-X., Yoshitake, T., Zhang, X.-L., Miry, O., Kehr, J., Stanton, P. K., Gross, A. L., Burgdorf, J. S., Kroes, R. A., & Moskal, J. R. (2019). Positive N-Methyl-D-Aspartate Receptor Modulation by Rapastinel Promotes Rapid and Sustained Antidepressant-Like Effects. The International Journal of Neuropsychopharmacology, 22(3), 247–259. https://doi.org/10.1093/ijnp/pyy101
Dong, C., Zhang, J.-C., Yao, W., Ren, Q., Ma, M., Yang, C., Chaki, S., & Hashimoto, K. (2017). Rapid and Sustained Antidepressant Action of the mGlu2/3 Receptor Antagonist MGS0039 in the Social Defeat Stress Model: Comparison with Ketamine. The International Journal of Neuropsychopharmacology, 20(3), 228–236. https://doi.org/10.1093/ijnp/pyw089
Drevets, W. C., & Furey, M. L. (2010). Replication of scopolamine’s antidepressant efficacy in major depressive disorder: A randomized, placebo-controlled clinical trial. Biological Psychiatry, 67(5), 432–438. https://doi.org/10.1016/j.biopsych.2009.11.021
Driesen, N. R., McCarthy, G., Bhagwagar, Z., Bloch, M., Calhoun, V., D’Souza, D. C., Gueorguieva, R., He, G., Ramachandran, R., Suckow, R. F., Anticevic, A., Morgan, P. T., & Krystal, J. H. (2013). Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Molecular Psychiatry, 18(11), 1199–1204. https://doi.org/10.1038/mp.2012.194
D’Sa, C., & Duman, R. S. (2002). Antidepressants and neuroplasticity. Bipolar Disorders, 4(3), 183–194. https://doi.org/10.1034/j.1399-5618.2002.01203.x
Du, J., Suzuki, K., Wei, Y., Wang, Y., Blumenthal, R., Chen, Z., Falke, C., Zarate, C. A., & Manji, H. K. (2007). The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: Relationship to clinical effects in mood disorders. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 32(4), 793–802. https://doi.org/10.1038/sj.npp.1301178
Dualé, C., Sibaud, F., Guastella, V., Vallet, L., Gimbert, Y.-A., Taheri, H., Filaire, M., Schoeffler, P., & Dubray, C. (2009). Perioperative ketamine does not prevent chronic pain after thoracotomy. European Journal of Pain (London, England), 13(5), 497–505. https://doi.org/10.1016/j.ejpain.2008.06.013
Duman, R. S. (2018). Ketamine and rapid-acting antidepressants: A new era in the battle against depression and suicide. F1000Research, 7. https://doi.org/10.12688/f1000research.14344.1
Duman, R. S., Heninger, G., & Nestler, E. (1997). A Molecular and Cellular Theory of Depression. Archives of General Psychiatry, 54(7), 597. https://doi.org/10.1001/archpsyc.1997.01830190015002
Duman, R. S., Li, N., Liu, R.-J., Duric, V., & Aghajanian, G. (2012). Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology, 62(1), 35–41. https://doi.org/10.1016/j.neuropharm.2011.08.044
Duncan, W. C., Sarasso, S., Ferrarelli, F., Selter, J., Riedner, B. A., Hejazi, N. S., Yuan, P., Brutsche, N., Manji, H. K., Tononi, G., & Zarate, C. A. (2013). Concomitant BDNF and sleep slow wave changes indicate ketamine-induced plasticity in major depressive disorder. The International Journal of Neuropsychopharmacology, 16(2), 301–311. https://doi.org/10.1017/S1461145712000545
Duric, V., Banasr, M., Stockmeier, C. A., Simen, A. A., Newton, S. S., Overholser, J. C., Jurjus, G. J., Dieter, L., & Duman, R. S. (2013). Altered expression of synapse and glutamate related genes in post-mortem hippocampus of depressed subjects. The International Journal of Neuropsychopharmacology, 16(1), 69–82. https://doi.org/10.1017/S1461145712000016
Dwyer, J. M., Lepack, A. E., & Duman, R. S. (2012). MTOR activation is required for the antidepressant effects of mGluR₂/₃ blockade. The International Journal of Neuropsychopharmacology, 15(4), 429–434. https://doi.org/10.1017/S1461145711001702
Ebert, B., Mikkelsen, S., Thorkildsen, C., & Borgbjerg, F. M. (1997). Norketamine, the main metabolite of ketamine, is a non-competitive NMDA receptor antagonist in the rat cortex and spinal cord. European Journal of Pharmacology, 333(1), 99–104. https://doi.org/10.1016/S0014-2999(97)01116-3
El Iskandrani, K. S., Oosterhof, C. A., El Mansari, M., & Blier, P. (2015). Impact of subanesthetic doses of ketamine on AMPA-mediated responses in rats: An in vivo electrophysiological study on monoaminergic and glutamatergic neurons. Journal of Psychopharmacology (Oxford, England), 29(7), 792–801. https://doi.org/10.1177/0269881115573809
Elizalde, N., García-García, A. L., Totterdell, S., Gendive, N., Venzala, E., Ramirez, M. J., Del Rio, J., & Tordera, R. M. (2010). Sustained stress-induced changes in mice as a model for chronic depression. Psychopharmacology, 210(3), 393–406. https://doi.org/10.1007/s00213-010-1835-6
Engin, E., Treit, D., & Dickson, C. T. (2009). Anxiolytic- and antidepressant-like properties of ketamine in behavioral and neurophysiological animal models. Neuroscience, 161(2), 359–369. https://doi.org/10.1016/j.neuroscience.2009.03.038
Erhardt, S., Lim, C. K., Linderholm, K. R., Janelidze, S., Lindqvist, D., Samuelsson, M., Lundberg, K., Postolache, T. T., Träskman-Bendz, L., Guillemin, G. J., & Brundin, L. (2013). Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 38(5), 743–752. https://doi.org/10.1038/npp.2012.248
Fatt, P., & Katz, B. (1952). Spontaneous subthreshold activity at motor nerve endings. The Journal of Physiology, 117(1), 109–128.
Fava, M., Memisoglu, A., Thase, M. E., Bodkin, J. A., Trivedi, M. H., de Somer, M., Du, Y., Leigh-Pemberton, R., DiPetrillo, L., Silverman, B., & Ehrich, E. (2016). Opioid Modulation With Buprenorphine/Samidorphan as Adjunctive Treatment for Inadequate Response to Antidepressants: A Randomized Double-Blind Placebo-Controlled Trial. The American Journal of Psychiatry, 173(5), 499–508. https://doi.org/10.1176/appi.ajp.2015.15070921
Featherstone, D. E., & Shippy, S. A. (2008). Regulation of synaptic transmission by ambient extracellular glutamate. The Neuroscientist: A Review Journal Bringing Neurobiology, Neurology and Psychiatry, 14(2), 171–181. https://doi.org/10.1177/1073858407308518
Featherstone, R. E., Liang, Y., Saunders, J. A., Tatard-Leitman, V. M., Ehrlichman, R. S., & Siegel, S. J. (2012). Subchronic ketamine treatment leads to permanent changes in EEG, cognition and the astrocytic glutamate transporter EAAT2 in mice. Neurobiology of Disease, 47(3), 338–346. https://doi.org/10.1016/j.nbd.2012.05.003
Feder, A., Parides, M. K., Murrough, J. W., Perez, A. M., Morgan, J. E., Saxena, S., Kirkwood, K., aan het Rot, M., Lapidus, K. A. B., Wan, L.-B., Iosifescu, D., & Charney, D. S. (2014). Efficacy of Intravenous Ketamine for Treatment of Chronic Posttraumatic Stress Disorder: A Randomized Clinical Trial. JAMA Psychiatry, 71(6), 681. https://doi.org/10.1001/jamapsychiatry.2014.62
Ferguson, J. M., & Shingleton, R. N. (2007). An open-label, flexible-dose study of memantine in major depressive disorder. Clinical Neuropharmacology, 30(3), 136–144. https://doi.org/10.1097/WNF.0b013e3180314ae7
Feyissa, A. M., Chandran, A., Stockmeier, C. A., & Karolewicz, B. (2009). Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 33(1), 70–75. https://doi.org/10.1016/j.pnpbp.2008.10.005
Finck, A. D., & Ngai, S. H. (1982). Opiate receptor mediation of ketamine analgesia. Anesthesiology, 56(4), 291–297. https://doi.org/10.1097/00000542-198204000-00011
Fischell, J., Van Dyke, A. M., Kvarta, M. D., LeGates, T. A., & Thompson, S. M. (2015). Rapid Antidepressant Action and Restoration of Excitatory Synaptic Strength After Chronic Stress by Negative Modulators of Alpha5-Containing GABAA Receptors. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 40(11), 2499–2509. https://doi.org/10.1038/npp.2015.112
Franceschelli, A., Sens, J., Herchick, S., Thelen, C., & Pitychoutis, P. M. (2015). Sex differences in the rapid and the sustained antidepressant-like effects of ketamine in stress-naïve and “depressed” mice exposed to chronic mild stress. Neuroscience, 290, 49–60. https://doi.org/10.1016/j.neuroscience.2015.01.008
Freland, L., & Beaulieu, J.-M. (2012). Inhibition of GSK3 by lithium, from single molecules to signaling networks. Frontiers in Molecular Neuroscience, 5, 14. https://doi.org/10.3389/fnmol.2012.00014
Fuchs, T., Jefferson, S. J., Hooper, A., Yee, P.-H., Maguire, J., & Luscher, B. (2017). Disinhibition of somatostatin-positive GABAergic interneurons results in an anxiolytic and antidepressant-like brain state. Molecular Psychiatry, 22(6), 920–930. https://doi.org/10.1038/mp.2016.188
Fukumoto, K., Fogaça, M. V., Liu, R.-J., Duman, C., Kato, T., Li, X.-Y., & Duman, R. S. (2019). Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketamine. Proceedings of the National Academy of Sciences, 116(1), 297–302. https://doi.org/10.1073/pnas.1814709116
Fukumoto, K., Iijima, M., & Chaki, S. (2014). Serotonin-1A receptor stimulation mediates effects of a metabotropic glutamate 2/3 receptor antagonist, 2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl)propanoic acid (LY341495), and an N-methyl-D-aspartate receptor antagonist, ketamine, in the novelty-suppressed feeding test. Psychopharmacology, 231(11), 2291–2298. https://doi.org/10.1007/s00213-013-3378-0
Fukumoto, K., Iijima, M., & Chaki, S. (2016). The Antidepressant Effects of an mGlu2/3 Receptor Antagonist and Ketamine Require AMPA Receptor Stimulation in the mPFC and Subsequent Activation of the 5-HT Neurons in the DRN. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 41(4), 1046–1056. https://doi.org/10.1038/npp.2015.233
Fukumoto, K., Toki, H., Iijima, M., Hashihayata, T., Yamaguchi, J.-I., Hashimoto, K., & Chaki, S. (2017). Antidepressant Potential of (R)-Ketamine in Rodent Models: Comparison with (S)-Ketamine. The Journal of Pharmacology and Experimental Therapeutics, 361(1), 9–16. https://doi.org/10.1124/jpet.116.239228
Furey, M. L., & Drevets, W. C. (2006). Antidepressant efficacy of the antimuscarinic drug scopolamine: A randomized, placebo-controlled clinical trial. Archives of General Psychiatry, 63(10), 1121–1129. https://doi.org/10.1001/archpsyc.63.10.1121
Gao, Y., Romero-Aleshire, M. J., Cai, Q., Price, T. J., & Brooks, H. L. (2013). Rapamycin inhibition of mTORC1 reverses lithium-induced proliferation of renal collecting duct cells. American Journal of Physiology. Renal Physiology, 305(8), F1201-1208. https://doi.org/10.1152/ajprenal.00153.2013
Garcia, L. S. B., Comim, C. M., Valvassori, S. S., Réus, G. Z., Barbosa, L. M., Andreazza, A. C., Stertz, L., Fries, G. R., Gavioli, E. C., Kapczinski, F., & Quevedo, J. (2008). Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 32(1), 140–144. https://doi.org/10.1016/j.pnpbp.2007.07.027
Garcia, L. S. B., Comim, C. M., Valvassori, S. S., Réus, G. Z., Stertz, L., Kapczinski, F., Gavioli, E. C., & Quevedo, J. (2009). Ketamine treatment reverses behavioral and physiological alterations induced by chronic mild stress in rats. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 33(3), 450–455. https://doi.org/10.1016/j.pnpbp.2009.01.004
Garcia, L. S., Comim, C. M., Valvassori, S. S., Réus, G. Z., Andreazza, A. C., Stertz, L., Fries, G. R., Gavioli, E. C., Kapczinski, F., & Quevedo, J. (2008). Chronic administration of ketamine elicits antidepressant-like effects in rats without affecting hippocampal brain-derived neurotrophic factor protein levels. Basic & Clinical Pharmacology & Toxicology, 103(6), 502–506. https://doi.org/10.1111/j.1742-7843.2008.00210.x
Gaynes, B. N., Warden, D., Trivedi, M. H., Wisniewski, S. R., Fava, M., & Rush, A. J. (2009). What Did STAR∗D Teach Us? Results From a Large-Scale, Practical, Clinical Trial for Patients With Depression. PSYCHIATRIC SERVICES, 60(11), 7.
Getachew, B., Aubee, J. I., Schottenfeld, R. S., Csoka, A. B., Thompson, K. M., & Tizabi, Y. (2018). Ketamine interactions with gut-microbiota in rats: Relevance to its antidepressant and anti-inflammatory properties. BMC Microbiology, 18(1), 222. https://doi.org/10.1186/s12866-018-1373-7
Ghosal, S., Bang, E., Yue, W., Hare, B. D., Lepack, A. E., Girgenti, M. J., & Duman, R. S. (2018). Activity-Dependent Brain-Derived Neurotrophic Factor Release Is Required for the Rapid Antidepressant Actions of Scopolamine. Biological Psychiatry, 83(1), 29–37. https://doi.org/10.1016/j.biopsych.2017.06.017
Gideons, E. S., Kavalali, E. T., & Monteggia, L. M. (2014). Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proceedings of the National Academy of Sciences of the United States of America, 111(23), 8649–8654. https://doi.org/10.1073/pnas.1323920111
Gigliucci, V., O’Dowd, G., Casey, S., Egan, D., Gibney, S., & Harkin, A. (2013). Ketamine elicits sustained antidepressant-like activity via a serotonin-dependent mechanism. Psychopharmacology, 228(1), 157–166. https://doi.org/10.1007/s00213-013-3024-x
Glue, P., Gulati, A., Le Nedelec, M., & Duffull, S. (2011). Dose- and exposure-response to ketamine in depression. Biological Psychiatry, 70(4), e9-10; author reply e11-12. https://doi.org/10.1016/j.biopsych.2010.11.031
Glue, P., Medlicott, N. J., Harland, S., Neehoff, S., Anderson-Fahey, B., Le Nedelec, M., Gray, A., & McNaughton, N. (2017). Ketamine’s dose-related effects on anxiety symptoms in patients with treatment refractory anxiety disorders. Journal of Psychopharmacology (Oxford, England), 31(10), 1302–1305. https://doi.org/10.1177/0269881117705089
Glue, P., Neehoff, S. M., Medlicott, N. J., Gray, A., Kibby, G., & McNaughton, N. (2018). Safety and efficacy of maintenance ketamine treatment in patients with treatment-refractory generalised anxiety and social anxiety disorders. Journal of Psychopharmacology (Oxford, England), 32(6), 663–667. https://doi.org/10.1177/0269881118762073
Golden, S. A., Covington, H. E., Berton, O., & Russo, S. J. (2011). A standardized protocol for repeated social defeat stress in mice. Nature Protocols, 6(8), 1183–1191. https://doi.org/10.1038/nprot.2011.361
Gonzalez-Burgos, G., Cho, R. Y., & Lewis, D. A. (2015). Alterations in Cortical Network Oscillations and Parvalbumin Neurons in Schizophrenia. Biological Psychiatry, 77(12), 1031–1040. https://doi.org/10.1016/j.biopsych.2015.03.010
Grégoire, M.-C., MacLellan, D. L., & Finley, G. A. (2008). A pediatric case of ketamine-associated cystitis. Urology, 71(6), 1232–1233. https://doi.org/10.1016/j.urology.2007.11.141
Greifenstein, F. E., Devault, M., Yoshitake, J., & Gajewski, J. E. (1958). A study of a 1-aryl cyclo hexyl amine for anesthesia. Anesthesia and Analgesia, 37(5), 283–294.
Grieco, S. F., Velmeshev, D., Magistri, M., Eldar-Finkelman, H., Faghihi, M. A., Jope, R. S., & Beurel, E. (2017). Ketamine up-regulates a cluster of intronic miRNAs within the serotonin receptor 2C gene by inhibiting glycogen synthase kinase-3. The World Journal of Biological Psychiatry: The Official Journal of the World Federation of Societies of Biological Psychiatry, 18(6), 445–456. https://doi.org/10.1080/15622975.2016.1224927
Grigoriadis, S., & Robinson, G. E. (2007). Gender issues in depression. Annals of Clinical Psychiatry: Official Journal of the American Academy of Clinical Psychiatrists, 19(4), 247–255. https://doi.org/10.1080/10401230701653294
Grunebaum, M. F., Galfalvy, H. C., Choo, T.-H., Keilp, J. G., Moitra, V. K., Parris, M. S., Marver, J. E., Burke, A. K., Milak, M. S., Sublette, M. E., Oquendo, M. A., & Mann, J. J. (2018). Ketamine for Rapid Reduction of Suicidal Thoughts in Major Depression: A Midazolam-Controlled Randomized Clinical Trial. The American Journal of Psychiatry, 175(4), 327–335. https://doi.org/10.1176/appi.ajp.2017.17060647
Guidi, J., Tomba, E., & Fava, G. A. (2016). The Sequential Integration of Pharmacotherapy and Psychotherapy in the Treatment of Major Depressive Disorder: A Meta-Analysis of the Sequential Model and a Critical Review of the Literature. The American Journal of Psychiatry, 173(2), 128–137. https://doi.org/10.1176/appi.ajp.2015.15040476
Gupta, A., Devi, L. A., & Gomes, I. (2011). Potentiation of μ-opioid receptor-mediated signaling by ketamine. Journal of Neurochemistry, 119(2), 294–302. https://doi.org/10.1111/j.1471-4159.2011.07361.x
Haile, C. N., Murrough, J. W., Iosifescu, D. V., Chang, L. C., Al Jurdi, R. K., Foulkes, A., Iqbal, S., Mahoney, J. J., De La Garza, R., Charney, D. S., Newton, T. F., & Mathew, S. J. (2014). Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. The International Journal of Neuropsychopharmacology, 17(2), 331–336. https://doi.org/10.1017/S1461145713001119
Hamilton, M. (1959). The assessment of anxiety states by rating. The British Journal of Medical Psychology, 32(1), 50–55. https://doi.org/10.1111/j.2044-8341.1959.tb00467.x
Hamilton, M. (1960). A rating scale for depression. Journal of Neurology, Neurosurgery, and Psychiatry, 23, 56–62. https://doi.org/10.1136/jnnp.23.1.56
Hanson, J. E., Weber, M., Meilandt, W. J., Wu, T., Luu, T., Deng, L., Shamloo, M., Sheng, M., Scearce-Levie, K., & Zhou, Q. (2013). GluN2B Antagonism Affects Interneurons and Leads to Immediate and Persistent Changes in Synaptic Plasticity, Oscillations, and Behavior. Neuropsychopharmacology, 38(7), 1221–1233. https://doi.org/10.1038/npp.2013.19
Harraz, M. M., Tyagi, R., Cortés, P., & Snyder, S. H. (2016). Antidepressant action of ketamine via mTOR is mediated by inhibition of nitrergic Rheb degradation. Molecular Psychiatry, 21(3), 313–319. https://doi.org/10.1038/mp.2015.211
Hashimoto, K. (2019). Rapid-acting antidepressant ketamine, its metabolites and other candidates: A historical overview and future perspective. Psychiatry and Clinical Neurosciences, 73(10), 613–627. https://doi.org/10.1111/pcn.12902
Hashimoto, K., Kakiuchi, T., Ohba, H., Nishiyama, S., & Tsukada, H. (2017). Reduction of dopamine D2/3 receptor binding in the striatum after a single administration of esketamine, but not R-ketamine: A PET study in conscious monkeys. European Archives of Psychiatry and Clinical Neuroscience, 267(2), 173–176. https://doi.org/10.1007/s00406-016-0692-7
Hasler, G., van der Veen, J. W., Tumonis, T., Meyers, N., Shen, J., & Drevets, W. C. (2007). Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Archives of General Psychiatry, 64(2), 193–200. https://doi.org/10.1001/archpsyc.64.2.193
Hasselbalch, B. J., Knorr, U., Hasselbalch, S. G., Gade, A., & Kessing, L. V. (2012). Cognitive deficits in the remitted state of unipolar depressive disorder. Neuropsychology, 26(5), 642–651. https://doi.org/10.1037/a0029301
Hasselbalch, B. J., Knorr, U., & Kessing, L. V. (2011). Cognitive impairment in the remitted state of unipolar depressive disorder: A systematic review. Journal of Affective Disorders, 134(1–3), 20–31. https://doi.org/10.1016/j.jad.2010.11.011
Heifets, B. D., Williams, N. R., Blasey, C., Sudheimer, K., Rodriguez, C. I., & Schatzberg, A. F. (2019). Interpreting Ketamine’s Opioid Receptor Dependent Effect: Response to Sanacora. The American Journal of Psychiatry, 176(3), 249–250. https://doi.org/10.1176/appi.ajp.2018.18091061r
Herrera-Guzmán, I., Gudayol-Ferré, E., Herrera-Abarca, J. E., Herrera-Guzmán, D., Montelongo-Pedraza, P., Padrós Blázquez, F., Peró-Cebollero, M., & Guàrdia-Olmos, J. (2010). Major Depressive Disorder in recovery and neuropsychological functioning: Effects of selective serotonin reuptake inhibitor and dual inhibitor depression treatments on residual cognitive deficits in patients with Major Depressive Disorder in recovery. Journal of Affective Disorders, 123(1–3), 341–350. https://doi.org/10.1016/j.jad.2009.10.009
Hertzberg, M. A., Butterfield, M. I., Feldman, M. E., Beckham, J. C., Sutherland, S. M., Connor, K. M., & Davidson, J. R. (1999). A preliminary study of lamotrigine for the treatment of posttraumatic stress disorder. Biological Psychiatry, 45(9), 1226–1229. https://doi.org/10.1016/s0006-3223(99)00011-6
Highland, J. N., Morris, P. J., Zanos, P., Lovett, J., Ghosh, S., Wang, A. Q., Zarate, C. A., Thomas, C. J., Moaddel, R., & Gould, T. D. (2019). Mouse, rat, and dog bioavailability and mouse oral antidepressant efficacy of (2R,6R)-hydroxynorketamine. Journal of Psychopharmacology (Oxford, England), 33(1), 12–24. https://doi.org/10.1177/0269881118812095
Hill, M. N., Hellemans, K. G. C., Verma, P., Gorzalka, B. B., & Weinberg, J. (2012). Neurobiology of chronic mild stress: Parallels to major depression. Neuroscience & Biobehavioral Reviews, 36(9), 2085–2117. https://doi.org/10.1016/j.neubiorev.2012.07.001
Hindmarch, I. (2001). Expanding the horizons of depression: Beyond the monoamine hypothesis. Human Psychopharmacology: Clinical and Experimental, 16(3), 203–218. https://doi.org/10.1002/hup.288
Hirschfeld, R. M. A. (2012). The Epidemiology of Depression and the Evolution of Treatment. The Journal of Clinical Psychiatry, 73(suppl 1), 5–9. https://doi.org/10.4088/JCP.11096su1c.01
Ho, C. C. K., Pezhman, H., Praveen, S., Goh, E. H., Lee, B. C., Zulkifli, M. Z., & Isa, M. R. (2010). Ketamine-associated ulcerative cystitis: A case report and literature review. The Malaysian Journal of Medical Sciences: MJMS, 17(2), 61–65.
Holi, M. M., Pelkonen, M., Karlsson, L., Kiviruusu, O., Ruuttu, T., Heilä, H., Tuisku, V., & Marttunen, M. (2005). Psychometric properties and clinical utility of the Scale for Suicidal Ideation (SSI) in adolescents. BMC Psychiatry, 5, 8. https://doi.org/10.1186/1471-244X-5-8
Hollis, F., & Kabbaj, M. (2014). Social Defeat as an Animal Model for Depression. ILAR Journal, 55(2), 221–232. https://doi.org/10.1093/ilar/ilu002
Hollon, S. D., DeRubeis, R. J., Shelton, R. C., Amsterdam, J. D., Salomon, R. M., O’Reardon, J. P., Lovett, M. L., Young, P. R., Haman, K. L., Freeman, B. B., & Gallop, R. (2005). Prevention of Relapse Following Cognitive Therapy vs Medications in Moderate to Severe Depression. Archives of General Psychiatry, 62(4), 417. https://doi.org/10.1001/archpsyc.62.4.417
Homayoun, H., & Moghaddam, B. (2007). NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 27(43), 11496–11500. https://doi.org/10.1523/JNEUROSCI.2213-07.2007
Horacek, J., Brunovsky, M., Novak, T., Tislerova, B., Palenicek, T., Bubenikova-Valesova, V., Spaniel, F., Koprivova, J., Mohr, P., Balikova, M., & Hoschl, C. (2010). Subanesthetic dose of ketamine decreases prefrontal theta cordance in healthy volunteers: Implications for antidepressant effect. Psychological Medicine, 40(9), 1443–1451. https://doi.org/10.1017/S0033291709991619
Hu, Y.-D., Xiang, Y.-T., Fang, J.-X., Zu, S., Sha, S., Shi, H., Ungvari, G. S., Correll, C. U., Chiu, H. F. K., Xue, Y., Tian, T.-F., Wu, A.-S., Ma, X., & Wang, G. (2016). Single i.v. ketamine augmentation of newly initiated escitalopram for major depression: Results from a randomized, placebo-controlled 4-week study. Psychological Medicine, 46(3), 623–635. https://doi.org/10.1017/S0033291715002159
Huang, N., Hua, D., Zhan, G., Li, S., Zhu, B., Jiang, R., Yang, L., Bi, J., Xu, H., Hashimoto, K., Luo, A., & Yang, C. (2019). Role of Actinobacteria and Coriobacteriia in the antidepressant effects of ketamine in an inflammation model of depression. Pharmacology, Biochemistry, and Behavior, 176, 93–100. https://doi.org/10.1016/j.pbb.2018.12.001
Ibrahim, L., Diazgranados, N., Franco-Chaves, J., Brutsche, N., Henter, I. D., Kronstein, P., Moaddel, R., Wainer, I., Luckenbaugh, D. A., Manji, H. K., & Zarate, C. A. (2012). Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: Results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 37(6), 1526–1533. https://doi.org/10.1038/npp.2011.338
Ionescu, D. F., Luckenbaugh, D. A., Niciu, M. J., Richards, E. M., & Zarate, C. A. (2015). A single infusion of ketamine improves depression scores in patients with anxious bipolar depression. Bipolar Disorders, 17(4), 438–443. https://doi.org/10.1111/bdi.12277
Irwin, S. A., & Iglewicz, A. (2010). Oral ketamine for the rapid treatment of depression and anxiety in patients receiving hospice care. Journal of Palliative Medicine, 13(7), 903–908. https://doi.org/10.1089/jpm.2010.9808
Irwin, S. A., Iglewicz, A., Nelesen, R. A., Lo, J. Y., Carr, C. H., Romero, S. D., & Lloyd, L. S. (2013). Daily oral ketamine for the treatment of depression and anxiety in patients receiving hospice care: A 28-day open-label proof-of-concept trial. Journal of Palliative Medicine, 16(8), 958–965. https://doi.org/10.1089/jpm.2012.0617
Jafarinia, M., Afarideh, M., Tafakhori, A., Arbabi, M., Ghajar, A., Noorbala, A. A., Saravi, M. A., Agah, E., & Akhondzadeh, S. (2016). Efficacy and safety of oral ketamine versus diclofenac to alleviate mild to moderate depression in chronic pain patients: A double-blind, randomized, controlled trial. Journal of Affective Disorders, 204, 1–8. https://doi.org/10.1016/j.jad.2016.05.076
Janelidze, S., Mattei, D., Westrin, Å., Träskman-Bendz, L., & Brundin, L. (2011). Cytokine levels in the blood may distinguish suicide attempters from depressed patients. Brain, Behavior, and Immunity, 25(2), 335–339. https://doi.org/10.1016/j.bbi.2010.10.010
Jevtovic-Todorovic, V., Benshoff, N., & Olney, J. W. (2000). Ketamine potentiates cerebrocortical damage induced by the common anaesthetic agent nitrous oxide in adult rats. British Journal of Pharmacology, 130(7), 1692–1698. https://doi.org/10.1038/sj.bjp.0703479
Jevtovic-Todorovic, Vesna, & Carter, L. B. (2005). The anesthetics nitrous oxide and ketamine are more neurotoxic to old than to young rat brain. Neurobiology of Aging, 26(6), 947–956. https://doi.org/10.1016/j.neurobiolaging.2004.07.009
Jiang, H., Ling, Z., Zhang, Y., Mao, H., Ma, Z., Yin, Y., Wang, W., Tang, W., Tan, Z., Shi, J., Li, L., & Ruan, B. (2015). Altered fecal microbiota composition in patients with major depressive disorder. Brain, Behavior, and Immunity, 48, 186–194. https://doi.org/10.1016/j.bbi.2015.03.016
Jick, H., Kaye, J. A., & Jick, S. S. (2004). Antidepressants and the Risk of Suicidal Behaviors. JAMA, 292(3), 338. https://doi.org/10.1001/jama.292.3.338
Kadriu, B., Gold, P. W., Luckenbaugh, D. A., Lener, M. S., Ballard, E. D., Niciu, M. J., Henter, I. D., Park, L. T., De Sousa, R. T., Yuan, P., Machado-Vieira, R., & Zarate, C. A. (2018). Acute ketamine administration corrects abnormal inflammatory bone markers in major depressive disorder. Molecular Psychiatry, 23(7), 1626–1631. https://doi.org/10.1038/mp.2017.109
Kalsi, S. S., Wood, D. M., & Dargan, P. I. (2011). The epidemiology and patterns of acute and chronic toxicity associated with recreational ketamine use. Emerging Health Threats Journal, 4, 7107. https://doi.org/10.3402/ehtj.v4i0.7107
Kanes, S., Colquhoun, H., Gunduz-Bruce, H., Raines, S., Arnold, R., Schacterle, A., Doherty, J., Epperson, C. N., Deligiannidis, K. M., Riesenberg, R., Hoffmann, E., Rubinow, D., Jonas, J., Paul, S., & Meltzer-Brody, S. (2017). Brexanolone (SAGE-547 injection) in post-partum depression: A randomised controlled trial. Lancet (London, England), 390(10093), 480–489. https://doi.org/10.1016/S0140-6736(17)31264-3
Kanes, S. J., Colquhoun, H., Doherty, J., Raines, S., Hoffmann, E., Rubinow, D. R., & Meltzer-Brody, S. (2017). Open-label, proof-of-concept study of brexanolone in the treatment of severe postpartum depression. Human Psychopharmacology, 32(2). https://doi.org/10.1002/hup.2576
Kang, H. J., Voleti, B., Hajszan, T., Rajkowska, G., Stockmeier, C. A., Licznerski, P., Lepack, A., Majik, M. S., Jeong, L. S., Banasr, M., Son, H., & Duman, R. S. (2012). Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nature Medicine, 18(9), 1413–1417. https://doi.org/10.1038/nm.2886
Kantrowitz, J. T., Halberstam, B., & Gangwisch, J. (2015). Single-dose ketamine followed by daily D-Cycloserine in treatment-resistant bipolar depression. The Journal of Clinical Psychiatry, 76(6), 737–738. https://doi.org/10.4088/JCP.14l09527
Kapur, S., & Seeman, P. (2001). Ketamine has equal affinity for NMDA receptors and the high-affinity state of the dopamine D2 receptor. Biological Psychiatry, 49(11), 954–957. https://doi.org/10.1016/s0006-3223(01)01110-6
Kapur, S., & Seeman, P. (2002). NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D(2) and serotonin 5-HT(2)receptors-implications for models of schizophrenia. Molecular Psychiatry, 7(8), 837–844. https://doi.org/10.1038/sj.mp.4001093
Kara, N. Z., Stukalin, Y., & Einat, H. (2018). Revisiting the validity of the mouse forced swim test: Systematic review and meta-analysis of the effects of prototypic antidepressants. Neuroscience & Biobehavioral Reviews, 84, 1–11. https://doi.org/10.1016/j.neubiorev.2017.11.003
Karasawa, J., Shimazaki, T., Kawashima, N., & Chaki, S. (2005). AMPA receptor stimulation mediates the antidepressant-like effect of a group II metabotropic glutamate receptor antagonist. Brain Research, 1042(1), 92–98. https://doi.org/10.1016/j.brainres.2005.02.032
Karch, D. L., Logan, J., McDaniel, D., Parks, S., Patel, N., & Centers for Disease Control and Prevention (CDC). (2012). Surveillance for violent deaths—National Violent Death Reporting System, 16 states, 2009. Morbidity and Mortality Weekly Report. Surveillance Summaries (Washington, D.C.: 2002), 61(6), 1–43.
Karege, F., Vaudan, G., Schwald, M., Perroud, N., & La Harpe, R. (2005). Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Research. Molecular Brain Research, 136(1–2), 29–37. https://doi.org/10.1016/j.molbrainres.2004.12.020
Karolewicz, B., Stockmeier, C. A., & Ordway, G. A. (2005). Elevated levels of the NR2C subunit of the NMDA receptor in the locus coeruleus in depression. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 30(8), 1557–1567. https://doi.org/10.1038/sj.npp.1300781
Kegeles, L. S., Abi-Dargham, A., Zea-Ponce, Y., Rodenhiser-Hill, J., Mann, J. J., Van Heertum, R. L., Cooper, T. B., Carlsson, A., & Laruelle, M. (2000). Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: Implications for schizophrenia. Biological Psychiatry, 48(7), 627–640. https://doi.org/10.1016/s0006-3223(00)00976-8
Kekesi, O., Tuboly, G., Szucs, M., Birkas, E., Morvay, Z., Benedek, G., & Horvath, G. (2011). Long-lasting, distinct changes in central opioid receptor and urinary bladder functions in models of schizophrenia in rats. European Journal of Pharmacology, 661(1–3), 35–41. https://doi.org/10.1016/j.ejphar.2011.04.022
Kessler, R. C., Chiu, W. T., Demler, O., & Walters, E. E. (2005). Prevalence, Severity, and Comorbidity of 12-Month DSM-IV Disorders in the National Comorbidity Survey Replication. Archives of General Psychiatry, 62(6), 617. https://doi.org/10.1001/archpsyc.62.6.617
Khlestova, E., Johnson, J. W., Krystal, J. H., & Lisman, J. (2016). The Role of GluN2C-Containing NMDA Receptors in Ketamine’s Psychotogenic Action and in Schizophrenia Models. The Journal of Neuroscience, 36(44), 11151–11157. https://doi.org/10.1523/JNEUROSCI.1203-16.2016
Kim, C. S., Brager, D. H., & Johnston, D. (2018). Perisomatic changes in h-channels regulate depressive behaviors following chronic unpredictable stress. Molecular Psychiatry, 23(4), 892–903. https://doi.org/10.1038/mp.2017.28
Kim, Chung Sub, Chang, P. Y., & Johnston, D. (2012). Enhancement of dorsal hippocampal activity by knockdown of HCN1 channels leads to anxiolytic- and antidepressant-like behaviors. Neuron, 75(3), 503–516. https://doi.org/10.1016/j.neuron.2012.05.027
Kim, D.-H., Sarbassov, D. D., Ali, S. M., King, J. E., Latek, R. R., Erdjument-Bromage, H., Tempst, P., & Sabatini, D. M. (2002). MTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex that Signals to the Cell Growth Machinery. Cell, 110(2), 163–175. https://doi.org/10.1016/S0092-8674(02)00808-5
Kinney, J. W., Davis, C., Tabarean, I., Conti, B., Bartfai, T., & Behrens, M. (2006). A Specific Role for NR2A-Containing NMDA Receptors in the Maintenance of Parvalbumin and GAD67 Immunoreactivity in Cultured Interneurons. Journal of Neuroscience, 26(5), 1604–1615. https://doi.org/10.1523/JNEUROSCI.4722-05.2006
Kishimoto, A., Kaneko, M., Gotoh, Y., & Hashimoto, K. (2012). Ifenprodil for the treatment of flashbacks in female posttraumatic stress disorder patients with a history of childhood sexual abuse. Biological Psychiatry, 71(4), e7-8. https://doi.org/10.1016/j.biopsych.2011.10.014
Kishimoto, T., Chawla, J. M., Hagi, K., Zarate, C. A., Kane, J. M., Bauer, M., & Correll, C. U. (2016). Single-dose infusion ketamine and non-ketamine N-methyl-d-aspartate receptor antagonists for unipolar and bipolar depression: A meta-analysis of efficacy, safety and time trajectories. Psychological Medicine, 46(7), 1459–1472. https://doi.org/10.1017/S0033291716000064
Klein, D. F. (2008). The Loss of Serendipity in Psychopharmacology. JAMA, 299(9), 1063. https://doi.org/10.1001/jama.299.9.1063
Koike, H., & Chaki, S. (2014). Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behavioural Brain Research, 271, 111–115. https://doi.org/10.1016/j.bbr.2014.05.065
Koike, H., Iijima, M., & Chaki, S. (2011a). Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behavioural Brain Research, 224(1), 107–111. https://doi.org/10.1016/j.bbr.2011.05.035
Koike, H., Iijima, M., & Chaki, S. (2011b). Involvement of the mammalian target of rapamycin signaling in the antidepressant-like effect of group II metabotropic glutamate receptor antagonists. Neuropharmacology, 61(8), 1419–1423. https://doi.org/10.1016/j.neuropharm.2011.08.034
Kollmar, R., Markovic, K., Thürauf, N., Schmitt, H., & Kornhuber, J. (2008). Ketamine followed by memantine for the treatment of major depression. The Australian and New Zealand Journal of Psychiatry, 42(2), 170. https://doi.org/10.1080/00048670701787628
Kotermanski, S. E., & Johnson, J. W. (2009). Mg2+ Imparts NMDA Receptor Subtype Selectivity to the Alzheimer’s Drug Memantine. Journal of Neuroscience, 29(9), 2774–2779. https://doi.org/10.1523/JNEUROSCI.3703-08.2009
Kovacsics, C. E., Gottesman, I. I., & Gould, T. D. (2009). Lithium’s antisuicidal efficacy: Elucidation of neurobiological targets using endophenotype strategies. Annual Review of Pharmacology and Toxicology, 49, 175–198. https://doi.org/10.1146/annurev.pharmtox.011008.145557
Kranaster, L., Kammerer-Ciernioch, J., Hoyer, C., & Sartorius, A. (2011). Clinically favourable effects of ketamine as an anaesthetic for electroconvulsive therapy: A retrospective study. European Archives of Psychiatry and Clinical Neuroscience, 261(8), 575–582. https://doi.org/10.1007/s00406-011-0205-7
Krystal, J. H., D’Souza, D. C., Karper, L. P., Bennett, A., Abi-Dargham, A., Abi-Saab, D., Cassello, K., Bowers, M. B., Vegso, S., Heninger, G. R., & Charney, D. S. (1999). Interactive effects of subanesthetic ketamine and haloperidol in healthy humans. Psychopharmacology, 145(2), 193–204. https://doi.org/10.1007/s002130051049
Krystal, J. H., Karper, L. P., Seibyl, J. P., Freeman, G. K., Delaney, R., Bremner, J. D., Heninger, G. R., Bowers, M. B., & Charney, D. S. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Archives of General Psychiatry, 51(3), 199–214. https://doi.org/10.1001/archpsyc.1994.03950030035004
Krystal, John H., Abi-Saab, W., Perry, E., D’Souza, D. C., Liu, N., Gueorguieva, R., McDougall, L., Hunsberger, T., Belger, A., Levine, L., & Breier, A. (2005). Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology, 179(1), 303–309. https://doi.org/10.1007/s00213-004-1982-8
Krystal, John H., Madonick, S., Perry, E., Gueorguieva, R., Brush, L., Wray, Y., Belger, A., & D’Souza, D. C. (2006). Potentiation of low dose ketamine effects by naltrexone: Potential implications for the pharmacotherapy of alcoholism. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 31(8), 1793–1800. https://doi.org/10.1038/sj.npp.1300994
Kudoh, A., Takahira, Y., Katagai, H., & Takazawa, T. (2002). Small-dose ketamine improves the postoperative state of depressed patients. Anesthesia and Analgesia, 95(1), 114–118, table of contents. https://doi.org/10.1097/00000539-200207000-00020
Lahti, A. C., Warfel, D., Michaelidis, T., Weiler, M. A., Frey, K., & Tamminga, C. A. (2001). Long-term outcome of patients who receive ketamine during research. Biological Psychiatry, 49(10), 869–875. https://doi.org/10.1016/s0006-3223(00)01037-4
Lahti, A., Koffel, B., LaPorte, D., & Tamminga, C. (1995). Subanesthetic Doses of Ketamine Stimulate Psychosis in Schizophrenia. Neuropsychopharmacology, 13(1), 9–19. https://doi.org/10.1016/0893-133X(94)00131-I
Lai, R., Katalinic, N., Glue, P., Somogyi, A. A., Mitchell, P. B., Leyden, J., Harper, S., & Loo, C. K. (2014). Pilot dose-response trial of i.v. Ketamine in treatment-resistant depression. The World Journal of Biological Psychiatry: The Official Journal of the World Federation of Societies of Biological Psychiatry, 15(7), 579–584. https://doi.org/10.3109/15622975.2014.922697
Laje, G., Lally, N., Mathews, D., Brutsche, N., Chemerinski, A., Akula, N., Kelmendi, B., Simen, A., McMahon, F. J., Sanacora, G., & Zarate, C. (2012). Brain-derived neurotrophic factor Val66Met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biological Psychiatry, 72(11), e27-28. https://doi.org/10.1016/j.biopsych.2012.05.031
Lally, N., Nugent, A. C., Luckenbaugh, D. A., Ameli, R., Roiser, J. P., & Zarate, C. A. (2014). Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Translational Psychiatry, 4, e469. https://doi.org/10.1038/tp.2014.105
Lally, Níall, Nugent, A. C., Luckenbaugh, D. A., Niciu, M. J., Roiser, J. P., & Zarate, C. A. (2015). Neural correlates of change in major depressive disorder anhedonia following open-label ketamine. Journal of Psychopharmacology (Oxford, England), 29(5), 596–607. https://doi.org/10.1177/0269881114568041
Lapidus, K. A. B., Levitch, C. F., Perez, A. M., Brallier, J. W., Parides, M. K., Soleimani, L., Feder, A., Iosifescu, D. V., Charney, D. S., & Murrough, J. W. (2014). A randomized controlled trial of intranasal ketamine in major depressive disorder. Biological Psychiatry, 76(12), 970–976. https://doi.org/10.1016/j.biopsych.2014.03.026
Lara, D. R., Bisol, L. W., & Munari, L. R. (2013). Antidepressant, mood stabilizing and procognitive effects of very low dose sublingual ketamine in refractory unipolar and bipolar depression. The International Journal of Neuropsychopharmacology, 16(9), 2111–2117. https://doi.org/10.1017/S1461145713000485
Lauterborn, J. C., Lynch, G., Vanderklish, P., Arai, A., & Gall, C. M. (2000). Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 20(1), 8–21.
Lenze, E. J., Skidmore, E. R., Begley, A. E., Newcomer, J. W., Butters, M. A., & Whyte, E. M. (2012). Memantine for late-life depression and apathy after a disabling medical event: A 12-week, double-blind placebo-controlled pilot study: Memantine for late-life depression. International Journal of Geriatric Psychiatry, 27(9), 974–980. https://doi.org/10.1002/gps.2813
Lepack, A. E., Bang, E., Lee, B., Dwyer, J. M., & Duman, R. S. (2016). Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology, 111, 242–252. https://doi.org/10.1016/j.neuropharm.2016.09.011
Lepack, A. E., Fuchikami, M., Dwyer, J. M., Banasr, M., & Duman, R. S. (2014). BDNF release is required for the behavioral actions of ketamine. The International Journal of Neuropsychopharmacology, 18(1). https://doi.org/10.1093/ijnp/pyu033
Lewis, D. A., & Moghaddam, B. (2006). Cognitive Dysfunction in Schizophrenia: Convergence of γ-Aminobutyric Acid and Glutamate Alterations. Archives of Neurology, 63(10), 1372. https://doi.org/10.1001/archneur.63.10.1372
Li, B., Piriz, J., Mirrione, M., Chung, C., Proulx, C. D., Schulz, D., Henn, F., & Malinow, R. (2011). Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature, 470(7335), 535–539. https://doi.org/10.1038/nature09742
Li, N., Lee, B., Liu, R.-J., Banasr, M., Dwyer, J. M., Iwata, M., Li, X.-Y., Aghajanian, G., & Duman, R. S. (2010). MTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science, 329(5994), 959–964. https://doi.org/10.1126/science.1190287
Li, N., Liu, R.-J., Dwyer, J. M., Banasr, M., Lee, B., Son, H., Li, X.-Y., Aghajanian, G., & Duman, R. S. (2011). Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biological Psychiatry, 69(8), 754–761. https://doi.org/10.1016/j.biopsych.2010.12.015
Li, X., Tizzano, J. P., Griffey, K., Clay, M., Lindstrom, T., & Skolnick, P. (2001). Antidepressant-like actions of an AMPA receptor potentiator (LY392098). Neuropharmacology, 40(8), 1028–1033. https://doi.org/10.1016/s0028-3908(00)00194-5
Li, Y., Zhu, Z. R., Ou, B. C., Wang, Y. Q., Tan, Z. B., Deng, C. M., Gao, Y. Y., Tang, M., So, J. H., Mu, Y. L., & Zhang, L. Q. (2015). Dopamine D2/D3 but not dopamine D1 receptors are involved in the rapid antidepressant-like effects of ketamine in the forced swim test. Behavioural Brain Research, 279, 100–105. https://doi.org/10.1016/j.bbr.2014.11.016
Liao, Y., Tang, J., Corlett, P. R., Wang, X., Yang, M., Chen, H., Liu, T., Chen, X., Hao, W., & Fletcher, P. C. (2011). Reduced dorsal prefrontal gray matter after chronic ketamine use. Biological Psychiatry, 69(1), 42–48. https://doi.org/10.1016/j.biopsych.2010.08.030
Liebrenz, M., Stohler, R., & Borgeat, A. (2009). Repeated intravenous ketamine therapy in a patient with treatment-resistant major depression. The World Journal of Biological Psychiatry: The Official Journal of the World Federation of Societies of Biological Psychiatry, 10(4 Pt 2), 640–643. https://doi.org/10.1080/15622970701420481
Lindqvist, D., Janelidze, S., Hagell, P., Erhardt, S., Samuelsson, M., Minthon, L., Hansson, O., Björkqvist, M., Träskman-Bendz, L., & Brundin, L. (2009). Interleukin-6 is elevated in the cerebrospinal fluid of suicide attempters and related to symptom severity. Biological Psychiatry, 66(3), 287–292. https://doi.org/10.1016/j.biopsych.2009.01.030
Liu, M.-Y., Yin, C.-Y., Zhu, L.-J., Zhu, X.-H., Xu, C., Luo, C.-X., Chen, H., Zhu, D.-Y., & Zhou, Q.-G. (2018). Sucrose preference test for measurement of stress-induced anhedonia in mice. Nature Protocols, 13(7), 1686–1698. https://doi.org/10.1038/s41596-018-0011-z
Liu, R.-J., Duman, C., Kato, T., Hare, B., Lopresto, D., Bang, E., Burgdorf, J., Moskal, J., Taylor, J., Aghajanian, G., & Duman, R. S. (2017). GLYX-13 Produces Rapid Antidepressant Responses with Key Synaptic and Behavioral Effects Distinct from Ketamine. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 42(6), 1231–1242. https://doi.org/10.1038/npp.2016.202
Liu, R.-J., Fuchikami, M., Dwyer, J. M., Lepack, A. E., Duman, R. S., & Aghajanian, G. K. (2013). GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 38(11), 2268–2277. https://doi.org/10.1038/npp.2013.128
Liu, R.-J., Lee, F. S., Li, X.-Y., Bambico, F., Duman, R. S., & Aghajanian, G. K. (2012). Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biological Psychiatry, 71(11), 996–1005. https://doi.org/10.1016/j.biopsych.2011.09.030
Liu, W.-X., Wang, J., Xie, Z.-M., Xu, N., Zhang, G.-F., Jia, M., Zhou, Z.-Q., Hashimoto, K., & Yang, J.-J. (2016). Regulation of glutamate transporter 1 via BDNF-TrkB signaling plays a role in the anti-apoptotic and antidepressant effects of ketamine in chronic unpredictable stress model of depression. Psychopharmacology, 233(3), 405–415. https://doi.org/10.1007/s00213-015-4128-2
Lofwall, M. R., Griffiths, R. R., & Mintzer, M. Z. (2006). Cognitive and subjective acute dose effects of intramuscular ketamine in healthy adults. Experimental and Clinical Psychopharmacology, 14(4), 439–449. https://doi.org/10.1037/1064-1297.14.4.439
Lommatzsch, M., Quarcoo, D., Schulte-Herbrüggen, O., Weber, H., Virchow, J. C., Renz, H., & Braun, A. (2005). Neurotrophins in murine viscera: A dynamic pattern from birth to adulthood. International Journal of Developmental Neuroscience: The Official Journal of the International Society for Developmental Neuroscience, 23(6), 495–500. https://doi.org/10.1016/j.ijdevneu.2005.05.009
Loo, C. K., Gálvez, V., O’Keefe, E., Mitchell, P. B., Hadzi-Pavlovic, D., Leyden, J., Harper, S., Somogyi, A. A., Lai, R., Weickert, C. S., & Glue, P. (2016). Placebo-controlled pilot trial testing dose titration and intravenous, intramuscular and subcutaneous routes for ketamine in depression. Acta Psychiatrica Scandinavica, 134(1), 48–56. https://doi.org/10.1111/acps.12572
Lorrain, D. S., Baccei, C. S., Bristow, L. J., Anderson, J. J., & Varney, M. A. (2003). Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: Modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience, 117(3), 697–706. https://doi.org/10.1016/s0306-4522(02)00652-8
Luckenbaugh, D. A., Ibrahim, L., Brutsche, N., Franco-Chaves, J., Mathews, D., Marquardt, C. A., Cassarly, C., & Zarate, C. A. (2012). Family history of alcohol dependence and antidepressant response to an N-methyl-D-aspartate antagonist in bipolar depression. Bipolar Disorders, 14(8), 880–887. https://doi.org/10.1111/bdi.12003
Luckenbaugh, D. A., Niciu, M. J., Ionescu, D. F., Nolan, N. M., Richards, E. M., Brutsche, N. E., Guevara, S., & Zarate, C. A. (2014). Do the dissociative side effects of ketamine mediate its antidepressant effects? Journal of Affective Disorders, 159, 56–61. https://doi.org/10.1016/j.jad.2014.02.017
Lukasiewicz, M., Gerard, S., Besnard, A., Falissard, B., Perrin, E., Sapin, H., Tohen, M., Reed, C., Azorin, J.-M., & The emblem study group. (2013). Young Mania Rating Scale: How to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort: YMRS severity cutoff. International Journal of Methods in Psychiatric Research, 22(1), 46–58. https://doi.org/10.1002/mpr.1379
Lumsden, E. W., Troppoli, T. A., Myers, S. J., Zanos, P., Aracava, Y., Kehr, J., Lovett, J., Kim, S., Wang, F.-H., Schmidt, S., Jenne, C. E., Yuan, P., Morris, P. J., Thomas, C. J., Zarate, C. A., Moaddel, R., Traynelis, S. F., Pereira, E. F. R., Thompson, S. M., … Gould, T. D. (2019). Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proceedings of the National Academy of Sciences, 116(11), 5160–5169. https://doi.org/10.1073/pnas.1816071116
Lutz, P.-E., & Kieffer, B. L. (2013). Opioid receptors: Distinct roles in mood disorders. Trends in Neurosciences, 36(3), 195–206. https://doi.org/10.1016/j.tins.2012.11.002
Ma, X.-C., Dang, Y.-H., Jia, M., Ma, R., Wang, F., Wu, J., Gao, C.-G., & Hashimoto, K. (2013). Long-lasting antidepressant action of ketamine, but not glycogen synthase kinase-3 inhibitor SB216763, in the chronic mild stress model of mice. PloS One, 8(2), e56053. https://doi.org/10.1371/journal.pone.0056053
Ma, Z., Zang, T., Birnbaum, S. G., Wang, Z., Johnson, J. E., Zhang, C.-L., & Parada, L. F. (2017). TrkB dependent adult hippocampal progenitor differentiation mediates sustained ketamine antidepressant response. Nature Communications, 8(1), 1668. https://doi.org/10.1038/s41467-017-01709-8
Macedo, D., Filho, A. J. M. C., Soares de Sousa, C. N., Quevedo, J., Barichello, T., Júnior, H. V. N., & Freitas de Lucena, D. (2017). Antidepressants, antimicrobials or both? Gut microbiota dysbiosis in depression and possible implications of the antimicrobial effects of antidepressant drugs for antidepressant effectiveness. Journal of Affective Disorders, 208, 22–32. https://doi.org/10.1016/j.jad.2016.09.012
Machado-Vieira, R., Gold, P. W., Luckenbaugh, D. A., Ballard, E. D., Richards, E. M., Henter, I. D., De Sousa, R. T., Niciu, M. J., Yuan, P., & Zarate, C. A. (2017). The role of adipokines in the rapid antidepressant effects of ketamine. Molecular Psychiatry, 22(1), 127–133. https://doi.org/10.1038/mp.2016.36
Maddox, V. H., Godefroi, E. F., & Parcell, R. F. (1965). The Synthesis of Phencyclidine and Other 1-Arylcyclohexylamines. Journal of Medicinal Chemistry, 8(2), 230–235. https://doi.org/10.1021/jm00326a019
Maeng, S., Zarate, C. A., Du, J., Schloesser, R. J., McCammon, J., Chen, G., & Manji, H. K. (2008). Cellular Mechanisms Underlying the Antidepressant Effects of Ketamine: Role of α-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionic Acid Receptors. Biological Psychiatry, 63(4), 349–352. https://doi.org/10.1016/j.biopsych.2007.05.028
Maes, M., Verkerk, R., Vandoolaeghe, E., Lin, A., & Scharpé, S. (1998). Serum levels of excitatory amino acids, serine, glycine, histidine, threonine, taurine, alanine and arginine in treatment-resistant depression: Modulation by treatment with antidepressants and prediction of clinical responsivity. Acta Psychiatrica Scandinavica, 97(4), 302–308. https://doi.org/10.1111/j.1600-0447.1998.tb10004.x
Malhotra, A. K., Adler, C. M., Kennison, S. D., Elman, I., Pickar, D., & Breier, A. (1997). Clozapine blunts N-methyl-D-aspartate antagonist-induced psychosis: A study with ketamine. Biological Psychiatry, 42(8), 664–668. https://doi.org/10.1016/s0006-3223(96)00546-x
Malhotra, A. K., Pinals, D. A., Adler, C. M., Elman, I., Clifton, A., Pickar, D., & Breier, A. (1997). Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology, 17(3), 141–150. https://doi.org/10.1016/S0893-133X(97)00036-5
Malhotra, A. K., Pinals, D. A., Weingartner, H., Sirocco, K., Missar, C. D., Pickar, D., & Breier, A. (1996). NMDA receptor function and human cognition: The effects of ketamine in healthy volunteers. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 14(5), 301–307. https://doi.org/10.1016/0893-133X(95)00137-3
Marcus, S. M., Young, E. A., Kerber, K. B., Kornstein, S., Farabaugh, A. H., Mitchell, J., Wisniewski, S. R., Balasubramani, G. K., Trivedi, M. H., & Rush, A. J. (2005). Gender differences in depression: Findings from the STAR*D study. Journal of Affective Disorders, 87(2–3), 141–150. https://doi.org/10.1016/j.jad.2004.09.008
Marland, S., Ellerton, J., Andolfatto, G., Strapazzon, G., Thomassen, O., Brandner, B., Weatherall, A., & Paal, P. (2013). Ketamine: Use in anesthesia. CNS Neuroscience & Therapeutics, 19(6), 381–389. https://doi.org/10.1111/cns.12072
Mathew, S. J., Murrough, J. W., aan het Rot, M., Collins, K. A., Reich, D. L., & Charney, D. S. (2010). Riluzole for relapse prevention following intravenous ketamine in treatment-resistant depression: A pilot randomized, placebo-controlled continuation trial. The International Journal of Neuropsychopharmacology, 13(1), 71–82. https://doi.org/10.1017/S1461145709000169
Mathisen, L. C., Skjelbred, P., Skoglund, L. A., & Oye, I. (1995). Effect of ketamine, an NMDA receptor inhibitor, in acute and chronic orofacial pain. Pain, 61(2), 215–220. https://doi.org/10.1016/0304-3959(94)00170-j
Matza, L. S., Morlock, R., Sexton, C., Malley, K., & Feltner, D. (2010). Identifying HAM-A cutoffs for mild, moderate, and severe generalized anxiety disorder. International Journal of Methods in Psychiatric Research, 19(4), 223–232. https://doi.org/10.1002/mpr.323
Mccarthy, D. A., Chen, G., Kaump, D. H., & Ensor, C. (1965). General Anesthetic and Other Pharmacological Properties of 2-(O-chlorophenyl)-2-methylamino cyclohexanone HCl (CI-58L). The Journal of New Drugs, 5(1), 21–33. https://doi.org/10.1002/j.1552-4604.1965.tb00219.x
McEwen, B. S. (1999). Stress and hippocampal plasticity. Annual Review of Neuroscience, 22, 105–122. https://doi.org/10.1146/annurev.neuro.22.1.105
McGhee, L. L., Maani, C. V., Garza, T. H., Gaylord, K. M., & Black, I. H. (2008). The correlation between ketamine and posttraumatic stress disorder in burned service members. The Journal of Trauma, 64(2 Suppl), S195-198; Discussion S197-198. https://doi.org/10.1097/TA.0b013e318160ba1d
McGirr, A., Berlim, M. T., Bond, D. J., Fleck, M. P., Yatham, L. N., & Lam, R. W. (2015). A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychological Medicine, 45(4), 693–704. https://doi.org/10.1017/S0033291714001603
McGirr, Alexander, Berlim, M. T., Bond, D. J., Neufeld, N. H., Chan, P. Y., Yatham, L. N., & Lam, R. W. (2015). A systematic review and meta-analysis of randomized controlled trials of adjunctive ketamine in electroconvulsive therapy: Efficacy and tolerability. Journal of Psychiatric Research, 62, 23–30. https://doi.org/10.1016/j.jpsychires.2015.01.003
McMakin, D. L., Olino, T. M., Porta, G., Dietz, L. J., Emslie, G., Clarke, G., Wagner, K. D., Asarnow, J. R., Ryan, N. D., Birmaher, B., Shamseddeen, W., Mayes, T., Kennard, B., Spirito, A., Keller, M., Lynch, F. L., Dickerson, J. F., & Brent, D. A. (2012). Anhedonia predicts poorer recovery among youth with selective serotonin reuptake inhibitor treatment-resistant depression. Journal of the American Academy of Child and Adolescent Psychiatry, 51(4), 404–411. https://doi.org/10.1016/j.jaac.2012.01.011
Mendola, C., Cammarota, G., Netto, R., Cecci, G., Pisterna, A., Ferrante, D., Casadio, C., & Della Corte, F. (2012). S(+)-ketamine for control of perioperative pain and prevention of post thoracotomy pain syndrome: A randomized, double-blind study. Minerva Anestesiologica, 78(7), 757–766.
Middela, S., & Pearce, I. (2011). Ketamine-induced vesicopathy: A literature review. International Journal of Clinical Practice, 65(1), 27–30. https://doi.org/10.1111/j.1742-1241.2010.02502.x
Milak, M. S., Proper, C. J., Mulhern, S. T., Parter, A. L., Kegeles, L. S., Ogden, R. T., Mao, X., Rodriguez, C. I., Oquendo, M. A., Suckow, R. F., Cooper, T. B., Keilp, J. G., Shungu, D. C., & Mann, J. J. (2016). A pilot in vivo proton magnetic resonance spectroscopy study of amino acid neurotransmitter response to ketamine treatment of major depressive disorder. Molecular Psychiatry, 21(3), 320–327. https://doi.org/10.1038/mp.2015.83
Miller, O. H., Moran, J. T., & Hall, B. J. (2016). Two cellular hypotheses explaining the initiation of ketamine’s antidepressant actions: Direct inhibition and disinhibition. Neuropharmacology, 100, 17–26. https://doi.org/10.1016/j.neuropharm.2015.07.028
Miller, O. H., Yang, L., Wang, C.-C., Hargroder, E. A., Zhang, Y., Delpire, E., & Hall, B. J. (2014). GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. ELife, 3, e03581. https://doi.org/10.7554/eLife.03581
Moaddel, R., Abdrakhmanova, G., Kozak, J., Jozwiak, K., Toll, L., Jimenez, L., Rosenberg, A., Tran, T., Xiao, Y., Zarate, C. A., & Wainer, I. W. (2013). Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholine-evoked currents in α7 nicotinic acetylcholine receptors. European Journal of Pharmacology, 698(1–3), 228–234. https://doi.org/10.1016/j.ejphar.2012.11.023
Moaddel, R., Luckenbaugh, D. A., Xie, Y., Villaseñor, A., Brutsche, N. E., Machado-Vieira, R., Ramamoorthy, A., Lorenzo, M. P., Garcia, A., Bernier, M., Torjman, M. C., Barbas, C., Zarate, C. A., & Wainer, I. W. (2015). D-serine plasma concentration is a potential biomarker of (R,S)-ketamine antidepressant response in subjects with treatment-resistant depression. Psychopharmacology, 232(2), 399–409. https://doi.org/10.1007/s00213-014-3669-0
Moaddel, R., Shardell, M., Khadeer, M., Lovett, J., Kadriu, B., Ravichandran, S., Morris, P. J., Yuan, P., Thomas, C. J., Gould, T. D., Ferrucci, L., & Zarate, C. A. (2018). Plasma metabolomic profiling of a ketamine and placebo crossover trial of major depressive disorder and healthy control subjects. Psychopharmacology, 235(10), 3017–3030. https://doi.org/10.1007/s00213-018-4992-7
Moaddel, R., Venkata, S. L. V., Tanga, M. J., Bupp, J. E., Green, C. E., Iyer, L., Furimsky, A., Goldberg, M. E., Torjman, M. C., & Wainer, I. W. (2010). A parallel chiral-achiral liquid chromatographic method for the determination of the stereoisomers of ketamine and ketamine metabolites in the plasma and urine of patients with complex regional pain syndrome. Talanta, 82(5), 1892–1904. https://doi.org/10.1016/j.talanta.2010.08.005
Moghaddam, B., Adams, B., Verma, A., & Daly, D. (1997). Activation of Glutamatergic Neurotransmission by Ketamine: A Novel Step in the Pathway from NMDA Receptor Blockade to Dopaminergic and Cognitive Disruptions Associated with the Prefrontal Cortex. The Journal of Neuroscience, 17(8), 2921–2927. https://doi.org/10.1523/JNEUROSCI.17-08-02921.1997
Molendijk, M. L., & de Kloet, E. R. (2019). Coping with the forced swim stressor: Current state-of-the-art. Behavioural Brain Research, 364, 1–10. https://doi.org/10.1016/j.bbr.2019.02.005
Monteggia, L. M., Barrot, M., Powell, C. M., Berton, O., Galanis, V., Gemelli, T., Meuth, S., Nagy, A., Greene, R. W., & Nestler, E. J. (2004). Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proceedings of the National Academy of Sciences, 101(29), 10827–10832. https://doi.org/10.1073/pnas.0402141101
Montgomery, S. A., & Asberg, M. (1979). A new depression scale designed to be sensitive to change. The British Journal of Psychiatry: The Journal of Mental Science, 134, 382–389. https://doi.org/10.1192/bjp.134.4.382
Morgan, C. J. A., Muetzelfeldt, L., & Curran, H. V. (2009). Ketamine use, cognition and psychological wellbeing: A comparison of frequent, infrequent and ex-users with polydrug and non-using controls. Addiction (Abingdon, England), 104(1), 77–87. https://doi.org/10.1111/j.1360-0443.2008.02394.x
Morgan, C. J. A., Muetzelfeldt, L., & Curran, H. V. (2010). Consequences of chronic ketamine self-administration upon neurocognitive function and psychological wellbeing: A 1-year longitudinal study. Addiction (Abingdon, England), 105(1), 121–133. https://doi.org/10.1111/j.1360-0443.2009.02761.x
Morrison, R. L., Fedgchin, M., Singh, J., Van Gerven, J., Zuiker, R., Lim, K. S., van der Ark, P., Wajs, E., Xi, L., Zannikos, P., & Drevets, W. C. (2018). Effect of intranasal esketamine on cognitive functioning in healthy participants: A randomized, double-blind, placebo-controlled study. Psychopharmacology, 235(4), 1107–1119. https://doi.org/10.1007/s00213-018-4828-5
Moskal, J. R., Burgdorf, J. S., Stanton, P. K., Kroes, R. A., Disterhoft, J. F., Burch, R. M., & Khan, M. A. (2017). The Development of Rapastinel (Formerly GLYX-13); A Rapid Acting and Long Lasting Antidepressant. Current Neuropharmacology, 15(1), 47–56. https://doi.org/10.2174/1570159×14666160321122703
Murray, C. J. L. (2013). The State of US Health, 1990-2010: Burden of Diseases, Injuries, and Risk Factors. JAMA, 310(6), 591. https://doi.org/10.1001/jama.2013.13805
Murrough, J. W., Soleimani, L., DeWilde, K. E., Collins, K. A., Lapidus, K. A., Iacoviello, B. M., Lener, M., Kautz, M., Kim, J., Stern, J. B., Price, R. B., Perez, A. M., Brallier, J. W., Rodriguez, G. J., Goodman, W. K., Iosifescu, D. V., & Charney, D. S. (2015). Ketamine for rapid reduction of suicidal ideation: A randomized controlled trial. Psychological Medicine, 45(16), 3571–3580. https://doi.org/10.1017/S0033291715001506
Murrough, James W., Burdick, K. E., Levitch, C. F., Perez, A. M., Brallier, J. W., Chang, L. C., Foulkes, A., Charney, D. S., Mathew, S. J., & Iosifescu, D. V. (2015). Neurocognitive effects of ketamine and association with antidepressant response in individuals with treatment-resistant depression: A randomized controlled trial. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 40(5), 1084–1090. https://doi.org/10.1038/npp.2014.298
Murrough, James W., Iosifescu, D. V., Chang, L. C., Al Jurdi, R. K., Green, C. E., Perez, A. M., Iqbal, S., Pillemer, S., Foulkes, A., Shah, A., Charney, D. S., & Mathew, S. J. (2013). Antidepressant efficacy of ketamine in treatment-resistant major depression: A two-site randomized controlled trial. The American Journal of Psychiatry, 170(10), 1134–1142. https://doi.org/10.1176/appi.ajp.2013.13030392
Murrough, James W., Perez, A. M., Pillemer, S., Stern, J., Parides, M. K., aan het Rot, M., Collins, K. A., Mathew, S. J., Charney, D. S., & Iosifescu, D. V. (2013). Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biological Psychiatry, 74(4), 250–256. https://doi.org/10.1016/j.biopsych.2012.06.022
Murrough, James W., Wan, L.-B., Iacoviello, B., Collins, K. A., Solon, C., Glicksberg, B., Perez, A. M., Mathew, S. J., Charney, D. S., Iosifescu, D. V., & Burdick, K. E. (2013). Neurocognitive effects of ketamine in treatment-resistant major depression: Association with antidepressant response. Psychopharmacology. https://doi.org/10.1007/s00213-013-3255-x
Nations, K. R., Dogterom, P., Bursi, R., Schipper, J., Greenwald, S., Zraket, D., Gertsik, L., Johnstone, J., Lee, A., Pande, Y., Ruigt, G., & Ereshefsky, L. (2012). Examination of Org 26576, an AMPA receptor positive allosteric modulator, in patients diagnosed with major depressive disorder: An exploratory, randomized, double-blind, placebo-controlled trial. Journal of Psychopharmacology (Oxford, England), 26(12), 1525–1539. https://doi.org/10.1177/0269881112458728
Nemeth, C. L., Paine, T. A., Rittiner, J. E., Béguin, C., Carroll, F. I., Roth, B. L., Cohen, B. M., & Carlezon, W. A. (2010). Role of kappa-opioid receptors in the effects of salvinorin A and ketamine on attention in rats. Psychopharmacology, 210(2), 263–274. https://doi.org/10.1007/s00213-010-1834-7
Neske, G. T., Patrick, S. L., & Connors, B. W. (2015). Contributions of Diverse Excitatory and Inhibitory Neurons to Recurrent Network Activity in Cerebral Cortex. Journal of Neuroscience, 35(3), 1089–1105. https://doi.org/10.1523/JNEUROSCI.2279-14.2015
Nguyen, L., Marshalek, P. J., Weaver, C. B., Cramer, K. J., Pollard, S. E., & Matsumoto, R. R. (2015). Off-label use of transmucosal ketamine as a rapid-acting antidepressant: A retrospective chart review. Neuropsychiatric Disease and Treatment, 11, 2667–2673. https://doi.org/10.2147/NDT.S88569
Nibuya, M., Morinobu, S., & Duman, R. S. (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 15(11), 7539–7547.
Niciu, M. J., Luckenbaugh, D. A., Ionescu, D. F., Guevara, S., Machado-Vieira, R., Richards, E. M., Brutsche, N. E., Nolan, N. M., & Zarate, C. A. (2014). Clinical predictors of ketamine response in treatment-resistant major depression. The Journal of Clinical Psychiatry, 75(5), e417-423. https://doi.org/10.4088/JCP.13m08698
Niciu, M. J., Luckenbaugh, D. A., Ionescu, D. F., Mathews, D. C., Richards, E. M., & Zarate, C. A. (2013). Subanesthetic dose ketamine does not induce an affective switch in three independent samples of treatment-resistant major depression. Biological Psychiatry, 74(10), e23-24. https://doi.org/10.1016/j.biopsych.2013.01.038
Niciu, M. J., Shovestul, B. J., Jaso, B. A., Farmer, C., Luckenbaugh, D. A., Brutsche, N. E., Park, L. T., Ballard, E. D., & Zarate, C. A. (2018). Features of dissociation differentially predict antidepressant response to ketamine in treatment-resistant depression. Journal of Affective Disorders, 232, 310–315. https://doi.org/10.1016/j.jad.2018.02.049
Nierenberg, A. A., Husain, M. M., Trivedi, M. H., Fava, M., Warden, D., Wisniewski, S. R., Miyahara, S., & Rush, A. J. (2010). Residual symptoms after remission of major depressive disorder with citalopram and risk of relapse: A STAR*D report. Psychological Medicine, 40(1), 41–50. https://doi.org/10.1017/S0033291709006011
Niesters, M., Martini, C., & Dahan, A. (2014). Ketamine for chronic pain: Risks and benefits. British Journal of Clinical Pharmacology, 77(2), 357–367. https://doi.org/10.1111/bcp.12094
Nishitani, N., Nagayasu, K., Asaoka, N., Yamashiro, M., Shirakawa, H., Nakagawa, T., & Kaneko, S. (2014). Raphe AMPA receptors and nicotinic acetylcholine receptors mediate ketamine-induced serotonin release in the rat prefrontal cortex. The International Journal of Neuropsychopharmacology, 17(8), 1321–1326. https://doi.org/10.1017/S1461145714000649
Noorzurani, R., Vicknasingam, B., & Narayanan, S. (2010). Illicit ketamine induced frequency of micturition in a young Malay woman. Drug and Alcohol Review, 29(3), 334–336. https://doi.org/10.1111/j.1465-3362.2009.00147.x
Nosyreva, E., Szabla, K., Autry, A. E., Ryazanov, A. G., Monteggia, L. M., & Kavalali, E. T. (2013). Acute suppression of spontaneous neurotransmission drives synaptic potentiation. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 33(16), 6990–7002. https://doi.org/10.1523/JNEUROSCI.4998-12.2013
Nowak, G., Li, Y., & Paul, I. A. (1996). Adaptation of cortical but not hippocampal NMDA receptors after chronic citalopram treatment. European Journal of Pharmacology, 295(1), 75–85. https://doi.org/10.1016/0014-2999(95)00585-4
Nowak, G., Trullas, R., Layer, R. T., Skolnick, P., & Paul, I. A. (1993). Adaptive changes in the N-methyl-D-aspartate receptor complex after chronic treatment with imipramine and 1-aminocyclopropanecarboxylic acid. The Journal of Pharmacology and Experimental Therapeutics, 265(3), 1380–1386.
Nowak, Gabriel, Ordway, G. A., & Paul, I. A. (1995). Alterations in the N-methyl-d-asparatate (NMDA) receptor complex in the frontal cortex of suicide victims. Brain Research, 675(1–2), 157–164. https://doi.org/10.1016/0006-8993(95)00057-W
Nugent, A. C., Diazgranados, N., Carlson, P. J., Ibrahim, L., Luckenbaugh, D. A., Brutsche, N., Herscovitch, P., Drevets, W. C., & Zarate, C. A. (2014). Neural correlates of rapid antidepressant response to ketamine in bipolar disorder. Bipolar Disorders, 16(2), 119–128. https://doi.org/10.1111/bdi.12118
Nugent, A. C., Robinson, S. E., Coppola, R., & Zarate, C. A. (2016). Preliminary differences in resting state MEG functional connectivity pre- and post-ketamine in major depressive disorder. Psychiatry Research. Neuroimaging, 254, 56–66. https://doi.org/10.1016/j.pscychresns.2016.06.006
O’Connor, R. M., Grenham, S., Dinan, T. G., & Cryan, J. F. (2013). microRNAs as novel antidepressant targets: Converging effects of ketamine and electroconvulsive shock therapy in the rat hippocampus. The International Journal of Neuropsychopharmacology, 16(8), 1885–1892. https://doi.org/10.1017/S1461145713000448
Okamoto, N., Nakai, T., Sakamoto, K., Nagafusa, Y., Higuchi, T., & Nishikawa, T. (2010). Rapid antidepressant effect of ketamine anesthesia during electroconvulsive therapy of treatment-resistant depression: Comparing ketamine and propofol anesthesia. The Journal of ECT, 26(3), 223–227. https://doi.org/10.1097/YCT.0b013e3181c3b0aa
Olney, J. W., Labruyere, J., & Price, M. T. (1989). Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science (New York, N.Y.), 244(4910), 1360–1362. https://doi.org/10.1126/science.2660263
Osby, U., Brandt, L., Correia, N., Ekbom, A., & Sparén, P. (2001). Excess mortality in bipolar and unipolar disorder in Sweden. Archives of General Psychiatry, 58(9), 844–850. https://doi.org/10.1001/archpsyc.58.9.844
Ostroff, R., Gonzales, M., & Sanacora, G. (2005). Antidepressant effect of ketamine during ECT. The American Journal of Psychiatry, 162(7), 1385–1386. https://doi.org/10.1176/appi.ajp.162.7.1385
Overall, J. E., & Gorham, D. R. (1962). The Brief Psychiatric Rating Scale. Psychological Reports, 10(3), 799–812. https://doi.org/10.2466/pr0.1962.10.3.799
Oye, I., Paulsen, O., & Maurset, A. (1992). Effects of ketamine on sensory perception: Evidence for a role of N-methyl-D-aspartate receptors. The Journal of Pharmacology and Experimental Therapeutics, 260(3), 1209–1213.
Pacheco, D. da F., Romero, T. R. L., & Duarte, I. D. G. (2014). Central antinociception induced by ketamine is mediated by endogenous opioids and μ- and δ-opioid receptors. Brain Research, 1562, 69–75. https://doi.org/10.1016/j.brainres.2014.03.026
Pan, W., Banks, W. A., Fasold, M. B., Bluth, J., & Kastin, A. J. (1998). Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology, 37(12), 1553–1561. https://doi.org/10.1016/s0028-3908(98)00141-5
Park, M., Newman, L. E., Gold, P. W., Luckenbaugh, D. A., Yuan, P., Machado-Vieira, R., & Zarate, C. A. (2017). Change in cytokine levels is not associated with rapid antidepressant response to ketamine in treatment-resistant depression. Journal of Psychiatric Research, 84, 113–118. https://doi.org/10.1016/j.jpsychires.2016.09.025
Parsons, C. G., Danysz, W., & Quack, G. (1999). Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist—A review of preclinical data. Neuropharmacology, 38(6), 735–767. https://doi.org/10.1016/s0028-3908(99)00019-2
Paul, I. A., Layer, R. T., Skolnick, P., & Nowak, G. (1993). Adaptation of the NMDA receptor in rat cortex following chronic electroconvulsive shock or imipramine. European Journal of Pharmacology: Molecular Pharmacology, 247(3), 305–311. https://doi.org/10.1016/0922-4106(93)90199-J
Paul, R. K., Singh, N. S., Khadeer, M., Moaddel, R., Sanghvi, M., Green, C. E., O’Loughlin, K., Torjman, M. C., Bernier, M., & Wainer, I. W. (2014). (R,S)-Ketamine metabolites (R,S)-norketamine and (2S,6S)-hydroxynorketamine increase the mammalian target of rapamycin function. Anesthesiology, 121(1), 149–159. https://doi.org/10.1097/ALN.0000000000000285
Paul, R., Schaaff, N., Padberg, F., Möller, H.-J., & Frodl, T. (2009). Comparison of racemic ketamine and S-ketamine in treatment-resistant major depression: Report of two cases. The World Journal of Biological Psychiatry: The Official Journal of the World Federation of Societies of Biological Psychiatry, 10(3), 241–244. https://doi.org/10.1080/15622970701714370
Pennybaker, S. J., Luckenbaugh, D. A., Park, L. T., Marquardt, C. A., & Zarate, C. A. (2017). Ketamine and Psychosis History: Antidepressant Efficacy and Psychotomimetic Effects Postinfusion. Biological Psychiatry, 82(5), e35–e36. https://doi.org/10.1016/j.biopsych.2016.08.041
Pennybaker, S. J., Niciu, M. J., Luckenbaugh, D. A., & Zarate, C. A. (2017). Symptomatology and predictors of antidepressant efficacy in extended responders to a single ketamine infusion. Journal of Affective Disorders, 208, 560–566. https://doi.org/10.1016/j.jad.2016.10.026
Permoda-Osip, A., Kisielewski, J., Bartkowska-Sniatkowska, A., & Rybakowski, J. K. (2015). Single ketamine infusion and neurocognitive performance in bipolar depression. Pharmacopsychiatry, 48(2), 78–79. https://doi.org/10.1055/s-0034-1394399
Perry, E. B., Cramer, J. A., Cho, H.-S., Petrakis, I. L., Karper, L. P., Genovese, A., O’Donnell, E., Krystal, J. H., D’Souza, D. C., & Yale Ketamine Study Group. (2007). Psychiatric safety of ketamine in psychopharmacology research. Psychopharmacology, 192(2), 253–260. https://doi.org/10.1007/s00213-007-0706-2
Persson, J., Hasselström, J., Maurset, A., Oye, I., Svensson, J. O., Almqvist, O., Scheinin, H., Gustafsson, L. L., & Almqvist, O. (2002). Pharmacokinetics and non-analgesic effects of S- and R-ketamines in healthy volunteers with normal and reduced metabolic capacity. European Journal of Clinical Pharmacology, 57(12), 869–875. https://doi.org/10.1007/s002280100353
Petralia, R. S., Wang, Y. X., Hua, F., Yi, Z., Zhou, A., Ge, L., Stephenson, F. A., & Wenthold, R. J. (2010). Organization of NMDA receptors at extrasynaptic locations. Neuroscience, 167(1), 68–87. https://doi.org/10.1016/j.neuroscience.2010.01.022
Pham, T. H., Mendez-David, I., Defaix, C., Guiard, B. P., Tritschler, L., David, D. J., & Gardier, A. M. (2017). Ketamine treatment involves medial prefrontal cortex serotonin to induce a rapid antidepressant-like activity in BALB/cJ mice. Neuropharmacology, 112(Pt A), 198–209. https://doi.org/10.1016/j.neuropharm.2016.05.010
Pham, Thu Ha, Defaix, C., Xu, X., Deng, S.-X., Fabresse, N., Alvarez, J.-C., Landry, D. W., Brachman, R. A., Denny, C. A., & Gardier, A. M. (2018). Common Neurotransmission Recruited in (R,S)-Ketamine and (2R,6R)-Hydroxynorketamine-Induced Sustained Antidepressant-like Effects. Biological Psychiatry, 84(1), e3–e6. https://doi.org/10.1016/j.biopsych.2017.10.020
Phelps, L. E., Brutsche, N., Moral, J. R., Luckenbaugh, D. A., Manji, H. K., & Zarate, C. A. (2009). Family history of alcohol dependence and initial antidepressant response to an N-methyl-D-aspartate antagonist. Biological Psychiatry, 65(2), 181–184. https://doi.org/10.1016/j.biopsych.2008.09.029
Pigott, H. E. (2015). The STAR*D Trial: It is Time to Reexamine the Clinical Beliefs That Guide the Treatment of Major Depression. The Canadian Journal of Psychiatry, 60(1), 9–13. https://doi.org/10.1177/070674371506000104
Pillai, A., Kale, A., Joshi, S., Naphade, N., Raju, M. S. V. K., Nasrallah, H., & Mahadik, S. P. (2010). Decreased BDNF levels in CSF of drug-naive first-episode psychotic subjects: Correlation with plasma BDNF and psychopathology. The International Journal of Neuropsychopharmacology, 13(4), 535–539. https://doi.org/10.1017/S1461145709991015
Pisoni, A., Strawbridge, R., Hodsoll, J., Powell, T. R., Breen, G., Hatch, S., Hotopf, M., Young, A. H., & Cleare, A. J. (2018). Growth Factor Proteins and Treatment-Resistant Depression: A Place on the Path to Precision. Frontiers in Psychiatry, 9, 386. https://doi.org/10.3389/fpsyt.2018.00386
Pizzagalli, D. A. (2014). Depression, stress, and anhedonia: Toward a synthesis and integrated model. Annual Review of Clinical Psychology, 10, 393–423. https://doi.org/10.1146/annurev-clinpsy-050212-185606
Poon, T. L., Wong, K. F., Chan, M. Y., Fung, K. W., Chu, S. K., Man, C. W., Yiu, M. K., & Leung, S. K. (2010). Upper gastrointestinal problems in inhalational ketamine abusers. Journal of Digestive Diseases, 11(2), 106–110. https://doi.org/10.1111/j.1751-2980.2010.00424.x
Popoli, M., Yan, Z., McEwen, B. S., & Sanacora, G. (2012). The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nature Reviews Neuroscience, 13(1), 22–37. https://doi.org/10.1038/nrn3138
Porsolt, R. D., Bertin, A., & Jalfre, M. (1978). “Behavioural despair” in rats and mice: Strain differences and the effects of imipramine. European Journal of Pharmacology, 51(3), 291–294. https://doi.org/10.1016/0014-2999(78)90414-4
Pothion, S., Bizot, J.-C., Trovero, F., & Belzung, C. (2004). Strain differences in sucrose preference and in the consequences of unpredictable chronic mild stress. Behavioural Brain Research, 155(1), 135–146. https://doi.org/10.1016/j.bbr.2004.04.008
Pozzi, L., Dorocic, I. P., Wang, X., Carlén, M., & Meletis, K. (2014). Mice Lacking NMDA Receptors in Parvalbumin Neurons Display Normal Depression-Related Behavior and Response to Antidepressant Action of NMDAR Antagonists. PLoS ONE, 9(1), e83879. https://doi.org/10.1371/journal.pone.0083879
Preskorn, S. H., Baker, B., Kolluri, S., Menniti, F. S., Krams, M., & Landen, J. W. (2008). An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. Journal of Clinical Psychopharmacology, 28(6), 631–637. https://doi.org/10.1097/JCP.0b013e31818a6cea
Preskorn, S., Macaluso, M., Mehra, D. O. V., Zammit, G., Moskal, J. R., Burch, R. M., & GLYX-13 Clinical Study Group. (2015). Randomized proof of concept trial of GLYX-13, an N-methyl-D-aspartate receptor glycine site partial agonist, in major depressive disorder nonresponsive to a previous antidepressant agent. Journal of Psychiatric Practice, 21(2), 140–149. https://doi.org/10.1097/01.pra.0000462606.17725.93
Price, R. B., Iosifescu, D. V., Murrough, J. W., Chang, L. C., Al Jurdi, R. K., Iqbal, S. Z., Soleimani, L., Charney, D. S., Foulkes, A. L., & Mathew, S. J. (2014). Effects of ketamine on explicit and implicit suicidal cognition: A randomized controlled trial in treatment-resistant depression. Depression and Anxiety, 31(4), 335–343. https://doi.org/10.1002/da.22253
Price, R. B., Nock, M. K., Charney, D. S., & Mathew, S. J. (2009). Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biological Psychiatry, 66(5), 522–526. https://doi.org/10.1016/j.biopsych.2009.04.029
Qu, Y., Yang, C., Ren, Q., Ma, M., Dong, C., & Hashimoto, K. (2017). Comparison of (R)-ketamine and lanicemine on depression-like phenotype and abnormal composition of gut microbiota in a social defeat stress model. Scientific Reports, 7(1), 15725. https://doi.org/10.1038/s41598-017-16060-7
Quirk, M. C., Sosulski, D., & Feierstein, C. (2009). A defined network of fast-spiking interneurons in orbitofrontal cortex: Responses to behavioral contingencies and ketamine administration. Frontiers in Systems Neuroscience, 3. https://doi.org/10.3389/neuro.06.013.2009
Rabiner, E. A. (2007). Imaging of striatal dopamine release elicited with NMDA antagonists: Is there anything there to be seen? Journal of Psychopharmacology (Oxford, England), 21(3), 253–258. https://doi.org/10.1177/0269881107077767
Rajkowska, G., Miguel-Hidalgo, J. J., Wei, J., Dilley, G., Pittman, S. D., Meltzer, H. Y., Overholser, J. C., Roth, B. L., & Stockmeier, C. A. (1999). Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biological Psychiatry, 45(9), 1085–1098. https://doi.org/10.1016/s0006-3223(99)00041-4
Rajkowska, Grazyna, Miguel-Hidalgo, J. J., Dubey, P., Stockmeier, C. A., & Krishnan, K. R. R. (2005). Prominent Reduction in Pyramidal Neurons Density in the Orbitofrontal Cortex of Elderly Depressed Patients. Biological Psychiatry, 58(4), 297–306. https://doi.org/10.1016/j.biopsych.2005.04.013
Rao, T. S., Kim, H. S., Lehmann, J., Martin, L. L., & Wood, P. L. (1989). Differential effects of phencyclidine (PCP) and ketamine on mesocortical and mesostriatal dopamine release in vivo. Life Sciences, 45(12), 1065–1072. https://doi.org/10.1016/0024-3205(89)90163-x
Rasmussen, K. G., Lineberry, T. W., Galardy, C. W., Kung, S., Lapid, M. I., Palmer, B. A., Ritter, M. J., Schak, K. M., Sola, C. L., Hanson, A. J., & Frye, M. A. (2013). Serial infusions of low-dose ketamine for major depression. Journal of Psychopharmacology (Oxford, England), 27(5), 444–450. https://doi.org/10.1177/0269881113478283
Raue, P. J., Schulberg, H. C., Heo, M., Klimstra, S., & Bruce, M. L. (2009). Patients’ depression treatment preferences and initiation, adherence, and outcome: A randomized primary care study. Psychiatric Services (Washington, D.C.), 60(3), 337–343. https://doi.org/10.1176/appi.ps.60.3.337
Ren, Z., Pribiag, H., Jefferson, S. J., Shorey, M., Fuchs, T., Stellwagen, D., & Luscher, B. (2016). Bidirectional Homeostatic Regulation of a Depression-Related Brain State by Gamma-Aminobutyric Acidergic Deficits and Ketamine Treatment. Biological Psychiatry, 80(6), 457–468. https://doi.org/10.1016/j.biopsych.2016.02.009
Réus, G. Z., Vieira, F. G., Abelaira, H. M., Michels, M., Tomaz, D. B., dos Santos, M. A. B., Carlessi, A. S., Neotti, M. V., Matias, B. I., Luz, J. R., Dal-Pizzol, F., & Quevedo, J. (2014). MAPK signaling correlates with the antidepressant effects of ketamine. Journal of Psychiatric Research, 55, 15–21. https://doi.org/10.1016/j.jpsychires.2014.04.010
Robson, M. J., Elliott, M., Seminerio, M. J., & Matsumoto, R. R. (2012). Evaluation of sigma (σ) receptors in the antidepressant-like effects of ketamine in vitro and in vivo. European Neuropsychopharmacology: The Journal of the European College of Neuropsychopharmacology, 22(4), 308–317. https://doi.org/10.1016/j.euroneuro.2011.08.002
Rodriguez, C. I., Kegeles, L. S., Flood, P., & Simpson, H. B. (2011). Rapid resolution of obsessions after an infusion of intravenous ketamine in a patient with treatment-resistant obsessive-compulsive disorder. The Journal of Clinical Psychiatry, 72(4), 567–569. https://doi.org/10.4088/JCP.10l06653
Rodriguez, C. I., Kegeles, L. S., Levinson, A., Feng, T., Marcus, S. M., Vermes, D., Flood, P., & Simpson, H. B. (2013). Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: Proof-of-concept. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 38(12), 2475–2483. https://doi.org/10.1038/npp.2013.150
Rodriguez, C. I., Wheaton, M., Zwerling, J., Steinman, S. A., Sonnenfeld, D., Galfalvy, H., & Simpson, H. B. (2016). Can exposure-based CBT extend the effects of intravenous ketamine in obsessive-compulsive disorder? An open-label trial. The Journal of Clinical Psychiatry, 77(3), 408–409. https://doi.org/10.4088/JCP.15l10138
Rosa, P. B., Neis, V. B., Ribeiro, C. M., Moretti, M., & Rodrigues, A. L. S. (2016). Antidepressant-like effects of ascorbic acid and ketamine involve modulation of GABAA and GABAB receptors. Pharmacological Reports: PR, 68(5), 996–1001. https://doi.org/10.1016/j.pharep.2016.05.010
Rowland, L. M., Bustillo, J. R., Mullins, P. G., Jung, R. E., Lenroot, R., Landgraf, E., Barrow, R., Yeo, R., Lauriello, J., & Brooks, W. M. (2005). Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: A 4-T proton MRS study. The American Journal of Psychiatry, 162(2), 394–396. https://doi.org/10.1176/appi.ajp.162.2.394
Rush, A. J., Trivedi, M. H., Wisniewski, S. R., Nierenberg, A. A., Stewart, J. W., Warden, D., Niederehe, G., Thase, M. E., Lavori, P. W., Lebowitz, B. D., McGrath, P. J., Rosenbaum, J. F., & Sackeim, H. A. (2006). Acute and Longer-Term Outcomes in Depressed Outpatients Requiring One or Several Treatment Steps: A STAR*D Report. Am J Psychiatry, 13.
Rybakowski, J. K., Permoda-Osip, A., Skibinska, M., Adamski, R., & Bartkowska-Sniatkowska, A. (2013). Single ketamine infusion in bipolar depression resistant to antidepressants: Are neurotrophins involved? Human Psychopharmacology, 28(1), 87–90. https://doi.org/10.1002/hup.2271
Ryves, W. J., & Harwood, A. J. (2001). Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochemical and Biophysical Research Communications, 280(3), 720–725. https://doi.org/10.1006/bbrc.2000.4169
Sałat, K., Siwek, A., Starowicz, G., Librowski, T., Nowak, G., Drabik, U., Gajdosz, R., & Popik, P. (2015). Antidepressant-like effects of ketamine, norketamine and dehydronorketamine in forced swim test: Role of activity at NMDA receptor. Neuropharmacology, 99, 301–307. https://doi.org/10.1016/j.neuropharm.2015.07.037
Saligan, L. N., Luckenbaugh, D. A., Slonena, E. E., Machado-Vieira, R., & Zarate, C. A. (2016). An assessment of the anti-fatigue effects of ketamine from a double-blind, placebo-controlled, crossover study in bipolar disorder. Journal of Affective Disorders, 194, 115–119. https://doi.org/10.1016/j.jad.2016.01.009
Salvadore, G., Cornwell, B. R., Colon-Rosario, V., Coppola, R., Grillon, C., Zarate, C. A., & Manji, H. K. (2009). Increased anterior cingulate cortical activity in response to fearful faces: A neurophysiological biomarker that predicts rapid antidepressant response to ketamine. Biological Psychiatry, 65(4), 289–295. https://doi.org/10.1016/j.biopsych.2008.08.014
Samuels, B. A., & Hen, R. (2011). Novelty-Suppressed Feeding in the Mouse. In T. D. Gould (Ed.), Mood and Anxiety Related Phenotypes in Mice (Vol. 63, pp. 107–121). Humana Press. https://doi.org/10.1007/978-1-61779-313-4_7
Samuels, B. A., Nautiyal, K. M., Kruegel, A. C., Levinstein, M. R., Magalong, V. M., Gassaway, M. M., Grinnell, S. G., Han, J., Ansonoff, M. A., Pintar, J. E., Javitch, J. A., Sames, D., & Hen, R. (2017). The Behavioral Effects of the Antidepressant Tianeptine Require the Mu-Opioid Receptor. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 42(10), 2052–2063. https://doi.org/10.1038/npp.2017.60
Sanacora, G, Smith, M. A., Pathak, S., Su, H.-L., Boeijinga, P. H., McCarthy, D. J., & Quirk, M. C. (2014). Lanicemine: A low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Molecular Psychiatry, 19(9), 978–985. https://doi.org/10.1038/mp.2013.130
Sanacora, Gerard. (2019). Caution Against Overinterpreting Opiate Receptor Stimulation as Mediating Antidepressant Effects of Ketamine. The American Journal of Psychiatry, 176(3), 249. https://doi.org/10.1176/appi.ajp.2018.18091061
Sarbassov, D. D., Ali, S. M., Kim, D.-H., Guertin, D. A., Latek, R. R., Erdjument-Bromage, H., Tempst, P., & Sabatini, D. M. (2004). Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current Biology, 14(14), 1296–1302. https://doi.org/10.1016/j.cub.2004.06.054
Sartorius, A., Kiening, K. L., Kirsch, P., von Gall, C. C., Haberkorn, U., Unterberg, A. W., Henn, F. A., & Meyer-Lindenberg, A. (2010). Remission of major depression under deep brain stimulation of the lateral habenula in a therapy-refractory patient. Biological Psychiatry, 67(2), e9–e11. https://doi.org/10.1016/j.biopsych.2009.08.027
Schak, K. M., Vande Voort, J. L., Johnson, E. K., Kung, S., Leung, J. G., Rasmussen, K. G., Palmer, B. A., & Frye, M. A. (2016). Potential Risks of Poorly Monitored Ketamine Use in Depression Treatment. The American Journal of Psychiatry, 173(3), 215–218. https://doi.org/10.1176/appi.ajp.2015.15081082
Scheidegger, M., Walter, M., Lehmann, M., Metzger, C., Grimm, S., Boeker, H., Boesiger, P., Henning, A., & Seifritz, E. (2012). Ketamine decreases resting state functional network connectivity in healthy subjects: Implications for antidepressant drug action. PloS One, 7(9), e44799. https://doi.org/10.1371/journal.pone.0044799
Schildkraut, J. J., & Kety, S. S. (1967). Biogenic Amines and Emotion. Science, 156(3771), 21–30. https://doi.org/10.1126/science.156.3771.21
Schobel, S. A., Chaudhury, N. H., Khan, U. A., Paniagua, B., Styner, M. A., Asllani, I., Inbar, B. P., Corcoran, C. M., Lieberman, J. A., Moore, H., & Small, S. A. (2013). Imaging Patients with Psychosis and a Mouse Model Establishes a Spreading Pattern of Hippocampal Dysfunction and Implicates Glutamate as a Driver. Neuron, 78(1), 81–93. https://doi.org/10.1016/j.neuron.2013.02.011
Schönenberg, M., Reichwald, U., Domes, G., Badke, A., & Hautzinger, M. (2005). Effects of peritraumatic ketamine medication on early and sustained posttraumatic stress symptoms in moderately injured accident victims. Psychopharmacology, 182(3), 420–425. https://doi.org/10.1007/s00213-005-0094-4
Segmiller, F., Rüther, T., Linhardt, A., Padberg, F., Berger, M., Pogarell, O., Möller, H.-J., Kohler, C., & Schüle, C. (2013). Repeated S-ketamine infusions in therapy resistant depression: A case series. Journal of Clinical Pharmacology, 53(9), 996–998. https://doi.org/10.1002/jcph.122
Seligman, M. E. P. (1972). Learned Helplessness. Annual Review of Medicine, 23(1), 407–412. https://doi.org/10.1146/annurev.me.23.020172.002203
Semkovska, M., & McLoughlin, D. M. (2010). Objective cognitive performance associated with electroconvulsive therapy for depression: A systematic review and meta-analysis. Biological Psychiatry, 68(6), 568–577. https://doi.org/10.1016/j.biopsych.2010.06.009
Shahani, R., Streutker, C., Dickson, B., & Stewart, R. J. (2007). Ketamine-associated ulcerative cystitis: A new clinical entity. Urology, 69(5), 810–812. https://doi.org/10.1016/j.urology.2007.01.038
Sharp, R. (2015). The Hamilton Rating Scale for Depression. Occupational Medicine, 65(4), 340–340. https://doi.org/10.1093/occmed/kqv043
Sheline, Y. I., Wang, P. W., Gado, M. H., Csernansky, J. G., & Vannier, M. W. (1996). Hippocampal atrophy in recurrent major depression. Proceedings of the National Academy of Sciences, 93(9), 3908–3913. https://doi.org/10.1073/pnas.93.9.3908
Sherwin, E., Sandhu, K. V., Dinan, T. G., & Cryan, J. F. (2016). May the Force Be With You: The Light and Dark Sides of the Microbiota–Gut–Brain Axis in Neuropsychiatry. CNS Drugs, 30(11), 1019–1041. https://doi.org/10.1007/s40263-016-0370-3
Shimizu, E., Hashimoto, K., & Iyo, M. (2004). Ethnic difference of the BDNF 196G/A (val66met) polymorphism frequencies: The possibility to explain ethnic mental traits. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics: The Official Publication of the International Society of Psychiatric Genetics, 126B(1), 122–123. https://doi.org/10.1002/ajmg.b.20118
Shirayama, Y., & Hashimoto, K. (2017). Effects of a single bilateral infusion of R-ketamine in the rat brain regions of a learned helplessness model of depression. European Archives of Psychiatry and Clinical Neuroscience, 267(2), 177–182. https://doi.org/10.1007/s00406-016-0718-1
Shirayama, Y., & Hashimoto, K. (2018). Lack of Antidepressant Effects of (2R,6R)-Hydroxynorketamine in a Rat Learned Helplessness Model: Comparison with (R)-Ketamine. The International Journal of Neuropsychopharmacology, 21(1), 84–88. https://doi.org/10.1093/ijnp/pyx108
Shiroma, P. R., Albott, C. S., Johns, B., Thuras, P., Wels, J., & Lim, K. O. (2014). Neurocognitive performance and serial intravenous subanesthetic ketamine in treatment-resistant depression. The International Journal of Neuropsychopharmacology, 17(11), 1805–1813. https://doi.org/10.1017/S1461145714001011
Shiroma, P. R., Johns, B., Kuskowski, M., Wels, J., Thuras, P., Albott, C. S., & Lim, K. O. (2014). Augmentation of response and remission to serial intravenous subanesthetic ketamine in treatment resistant depression. Journal of Affective Disorders, 155, 123–129. https://doi.org/10.1016/j.jad.2013.10.036
Short, B., Fong, J., Galvez, V., Shelker, W., & Loo, C. K. (2018). Side-effects associated with ketamine use in depression: A systematic review. The Lancet Psychiatry, 5(1), 65–78. https://doi.org/10.1016/S2215-0366(17)30272-9
Simon, G. E., Revicki, D., Heiligenstein, J., Grothaus, L., VonKorff, M., Katon, W. J., & Hylan, T. R. (2000). Recovery from depression, work productivity, and health care costs among primary care patients. General Hospital Psychiatry, 22(3), 153–162. https://doi.org/10.1016/s0163-8343(00)00072-4
Singh, J. B., Fedgchin, M., Daly, E., Xi, L., Melman, C., De Bruecker, G., Tadic, A., Sienaert, P., Wiegand, F., Manji, H., Drevets, W. C., & Van Nueten, L. (2016). Intravenous Esketamine in Adult Treatment-Resistant Depression: A Double-Blind, Double-Randomization, Placebo-Controlled Study. Biological Psychiatry, 80(6), 424–431. https://doi.org/10.1016/j.biopsych.2015.10.018
Skolnick, P., Layer, R., Popik, P., Nowak, G., Paul, I., & Trullas, R. (1996). Adaptation of N-Methyl-D-Aspartate (NMDA) Receptors following Antidepressant Treatment: Implications for the Pharmacotherapy of Depression. Pharmacopsychiatry, 29(01), 23–26. https://doi.org/10.1055/s-2007-979537
Slattery, D. A., & Cryan, J. F. (2012). Using the rat forced swim test to assess antidepressant-like activity in rodents. Nature Protocols, 7(6), 1009–1014. https://doi.org/10.1038/nprot.2012.044
Slattery, D. A., Neumann, I. D., & Cryan, J. F. (2011). Transient inactivation of the infralimbic cortex induces antidepressant-like effects in the rat. Journal of Psychopharmacology, 25(10), 1295–1303. https://doi.org/10.1177/0269881110368873
Smith, D., Bouchal, R., Desanctis, C., Monroe, P., Amedro, J., Perrotti, J., & Crisp, T. (1987). Properties of the interaction between ketamine and opiate binding sites in vivo and in vitro. Neuropharmacology, 26(9), 1253–1260. https://doi.org/10.1016/0028-3908(87)90084-0
Smith, D. J., Perrotti, J. M., Mansell, A. L., & Monroe, P. J. (1985). Ketamine analgesia is not related to an opiate action in the periaqueductal gray region of the rat brain. Pain, 21(3), 253–265. https://doi.org/10.1016/0304-3959(85)90089-2
Smith, E. G., Deligiannidis, K. M., Ulbricht, C. M., Landolin, C. S., Patel, J. K., & Rothschild, A. J. (2013). Antidepressant Augmentation Using the N-methyl-D-aspartate Antagonist Memantine: A Randomized, Double-Blind, Placebo-Controlled Trial. The Journal of Clinical Psychiatry, 74(10), 966–973. https://doi.org/10.4088/JCP.12m08252
Smith, G. S., Schloesser, R., Brodie, J. D., Dewey, S. L., Logan, J., Vitkun, S. A., Simkowitz, P., Hurley, A., Cooper, T., Volkow, N. D., & Cancro, R. (1998). Glutamate modulation of dopamine measured in vivo with positron emission tomography (PET) and 11C-raclopride in normal human subjects. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 18(1), 18–25. https://doi.org/10.1016/S0893-133X(97)00092-4
Sos, P., Klirova, M., Novak, T., Kohutova, B., Horacek, J., & Palenicek, T. (2013). Relationship of ketamine’s antidepressant and psychotomimetic effects in unipolar depression. Neuro Endocrinology Letters, 34(4), 287–293.
Spijker, J., Bijl, R. V., de Graaf, R., & Nolen, W. A. (2001). Determinants of poor 1-year outcome of DSM-III-R major depression in the general population: Results of the Netherlands Mental Health Survey and Incidence Study (NEMESIS). Acta Psychiatrica Scandinavica, 103(2), 122–130. https://doi.org/10.1034/j.1600-0447.2001.103002122.x
Spijker, Jan, de Graaf, R., Ten Have, M., Nolen, W. A., & Speckens, A. (2010). Predictors of suicidality in depressive spectrum disorders in the general population: Results of the Netherlands Mental Health Survey and Incidence Study. Social Psychiatry and Psychiatric Epidemiology, 45(5), 513–521. https://doi.org/10.1007/s00127-009-0093-6
Stanciu, C. N., Glass, O. M., & Penders, T. M. (2017). Use of Buprenorphine in treatment of refractory depression-A review of current literature. Asian Journal of Psychiatry, 26, 94–98. https://doi.org/10.1016/j.ajp.2017.01.015
Steru, L., Chermat, R., Thierry, B., Mico, J. A., Lenegre, A., Steru, M., Simon, P., & Porsolt, R. D. (1987). The automated Tail Suspension Test: A computerized device which differentiates psychotropic drugs. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 11(6), 659–671. https://doi.org/10.1016/0278-5846(87)90002-9
Steru, L., Chermat, R., Thierry, B., & Simon, P. (1985). The tail suspension test: A new method for screening antidepressants in mice. Psychopharmacology, 85(3), 367–370. https://doi.org/10.1007/BF00428203
Stone, J. M., Dietrich, C., Edden, R., Mehta, M. A., De Simoni, S., Reed, L. J., Krystal, J. H., Nutt, D., & Barker, G. J. (2012). Ketamine effects on brain GABA and glutamate levels with 1H-MRS: Relationship to ketamine-induced psychopathology. Molecular Psychiatry, 17(7), 664–665. https://doi.org/10.1038/mp.2011.171
Storr, T. M., & Quibell, R. (2009). Can ketamine prescribed for pain cause damage to the urinary tract? Palliative Medicine, 23(7), 670–672. https://doi.org/10.1177/0269216309106828
Su, T.-P., Chen, M.-H., Li, C.-T., Lin, W.-C., Hong, C.-J., Gueorguieva, R., Tu, P.-C., Bai, Y.-M., Cheng, C.-M., & Krystal, J. H. (2017). Dose-Related Effects of Adjunctive Ketamine in Taiwanese Patients with Treatment-Resistant Depression. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 42(13), 2482–2492. https://doi.org/10.1038/npp.2017.94
Sun, H.-L., Zhou, Z.-Q., Zhang, G.-F., Yang, C., Wang, X.-M., Shen, J.-C., Hashimoto, K., & Yang, J.-J. (2016). Role of hippocampal p11 in the sustained antidepressant effect of ketamine in the chronic unpredictable mild stress model. Translational Psychiatry, 6, e741. https://doi.org/10.1038/tp.2016.21
Sutton, M. A., Ito, H. T., Cressy, P., Kempf, C., Woo, J. C., & Schuman, E. M. (2006). Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell, 125(4), 785–799. https://doi.org/10.1016/j.cell.2006.03.040
Sutton, M. A., Taylor, A. M., Ito, H. T., Pham, A., & Schuman, E. M. (2007). Postsynaptic decoding of neural activity: EEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron, 55(4), 648–661. https://doi.org/10.1016/j.neuron.2007.07.030
Sutton, M. A., Wall, N. R., Aakalu, G. N., & Schuman, E. M. (2004). Regulation of dendritic protein synthesis by miniature synaptic events. Science (New York, N.Y.), 304(5679), 1979–1983. https://doi.org/10.1126/science.1096202
Suzuki, K., Nosyreva, E., Hunt, K. W., Kavalali, E. T., & Monteggia, L. M. (2017). Effects of a ketamine metabolite on synaptic NMDAR function. Nature, 546(7659), E1–E3. https://doi.org/10.1038/nature22084
Svenningsson, P., Kim, Y., Warner-Schmidt, J., Oh, Y.-S., & Greengard, P. (2013). P11 and its role in depression and therapeutic responses to antidepressants. Nature Reviews. Neuroscience, 14(10), 673–680. https://doi.org/10.1038/nrn3564
Tam, Y.-H., Ng, C.-F., Pang, K. K.-Y., Yee, C.-H., Chu, W. C.-W., Leung, V. Y.-F., Wong, G. L.-H., Wong, V. W.-S., Chan, H. L.-Y., & Lai, P. B.-S. (2014). One-stop clinic for ketamine-associated uropathy: Report on service delivery model, patients’ characteristics and non-invasive investigations at baseline by a cross-sectional study in a prospective cohort of 318 teenagers and young adults. BJU International, 114(5), 754–760. https://doi.org/10.1111/bju.12675
Tang, J., Xue, W., Xia, B., Ren, L., Tao, W., Chen, C., Zhang, H., Wu, R., Wang, Q., Wu, H., Duan, J., & Chen, G. (2015). Involvement of normalized NMDA receptor and mTOR-related signaling in rapid antidepressant effects of Yueju and ketamine on chronically stressed mice. Scientific Reports, 5, 13573. https://doi.org/10.1038/srep13573
Tang, W. K., Liang, H. J., Lau, C. G., Tang, A., & Ungvari, G. S. (2013). Relationship Between Cognitive Impairment and Depressive Symptoms in Current Ketamine Users. Journal of Studies on Alcohol and Drugs, 74(3), 460–468. https://doi.org/10.15288/jsad.2013.74.460
Taylor, J. H., Landeros-Weisenberger, A., Coughlin, C., Mulqueen, J., Johnson, J. A., Gabriel, D., Reed, M. O., Jakubovski, E., & Bloch, M. H. (2018). Ketamine for Social Anxiety Disorder: A Randomized, Placebo-Controlled Crossover Trial. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 43(2), 325–333. https://doi.org/10.1038/npp.2017.194
Taylor, S. M. (2008). Electroconvulsive therapy, brain-derived neurotrophic factor, and possible neurorestorative benefit of the clinical application of electroconvulsive therapy. The Journal of ECT, 24(2), 160–165. https://doi.org/10.1097/YCT.0b013e3181571ad0
Teixeira, A. L., Barbosa, I. G., Diniz, B. S., & Kummer, A. (2010). Circulating levels of brain-derived neurotrophic factor: Correlation with mood, cognition and motor function. Biomarkers in Medicine, 4(6), 871–887. https://doi.org/10.2217/bmm.10.111
Thelen, C., Sens, J., Mauch, J., Pandit, R., & Pitychoutis, P. M. (2016). Repeated ketamine treatment induces sex-specific behavioral and neurochemical effects in mice. Behavioural Brain Research, 312, 305–312. https://doi.org/10.1016/j.bbr.2016.06.041
Tian, Z., Dong, C., Fujita, A., Fujita, Y., & Hashimoto, K. (2018). Expression of heat shock protein HSP-70 in the retrosplenial cortex of rat brain after administration of (R,S)-ketamine and (S)-ketamine, but not (R)-ketamine. Pharmacology, Biochemistry, and Behavior, 172, 17–21. https://doi.org/10.1016/j.pbb.2018.07.003
Tian, Z., Dong, C., Zhang, K., Chang, L., & Hashimoto, K. (2018). Lack of Antidepressant Effects of Low-Voltage-Sensitive T-Type Calcium Channel Blocker Ethosuximide in a Chronic Social Defeat Stress Model: Comparison with (R)-Ketamine. The International Journal of Neuropsychopharmacology, 21(11), 1031–1036. https://doi.org/10.1093/ijnp/pyy072
Tizabi, Y., Bhatti, B. H., Manaye, K. F., Das, J. R., & Akinfiresoye, L. (2012). Antidepressant-like effects of low ketamine dose is associated with increased hippocampal AMPA/NMDA receptor density ratio in female Wistar-Kyoto rats. Neuroscience, 213, 72–80. https://doi.org/10.1016/j.neuroscience.2012.03.052
Toth, E., Gersner, R., Wilf-Yarkoni, A., Raizel, H., Dar, D. E., Richter-Levin, G., Levit, O., & Zangen, A. (2008). Age-dependent effects of chronic stress on brain plasticity and depressive behavior. Journal of Neurochemistry, 107(2), 522–532. https://doi.org/10.1111/j.1471-4159.2008.05642.x
Trivedi, M. H., Rush, A. J., Wisniewski, S. R., Nierenberg, A. A., Warden, D., Ritz, L., Norquist, G., Howland, R. H., Lebowitz, B., McGrath, P. J., Shores-Wilson, K., Biggs, M. M., Balasubramani, G. K., Fava, M., & STAR*D Study Team. (2006). Evaluation of Outcomes With Citalopram for Depression Using Measurement-Based Care in STAR*D: Implications for Clinical Practice. American Journal of Psychiatry, 163(1), 28–40. https://doi.org/10.1176/appi.ajp.163.1.28
Trullas, R., & Skolnick, P. (1990). Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. European Journal of Pharmacology, 185(1), 1–10. https://doi.org/10.1016/0014-2999(90)90204-J
Tyler, M. W., Yourish, H. B., Ionescu, D. F., & Haggarty, S. J. (2017). Classics in Chemical Neuroscience: Ketamine. ACS Chemical Neuroscience, 8(6), 1122–1134. https://doi.org/10.1021/acschemneuro.7b00074
Undurraga, J., & Baldessarini, R. J. (2012). Randomized, Placebo-Controlled Trials of Antidepressants for Acute Major Depression: Thirty-Year Meta-Analytic Review. Neuropsychopharmacology, 37(4), 851–864. https://doi.org/10.1038/npp.2011.306
Valentine, G. W., Mason, G. F., Gomez, R., Fasula, M., Watzl, J., Pittman, B., Krystal, J. H., & Sanacora, G. (2011). The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [(1)H]-MRS. Psychiatry Research, 191(2), 122–127. https://doi.org/10.1016/j.pscychresns.2010.10.009
van de Loo, A. J. A. E., Bervoets, A. C., Mooren, L., Bouwmeester, N. H., Garssen, J., Zuiker, R., van Amerongen, G., van Gerven, J., Singh, J., der Ark, P. V., Fedgchin, M., Morrison, R., Wajs, E., & Verster, J. C. (2017). The effects of intranasal esketamine (84 mg) and oral mirtazapine (30 mg) on on-road driving performance: A double-blind, placebo-controlled study. Psychopharmacology, 234(21), 3175–3183. https://doi.org/10.1007/s00213-017-4706-6
Vande Voort, J. L., Morgan, R. J., Kung, S., Rasmussen, K. G., Rico, J., Palmer, B. A., Schak, K. M., Tye, S. J., Ritter, M. J., Frye, M. A., & Bobo, W. V. (2016). Continuation phase intravenous ketamine in adults with treatment-resistant depression. Journal of Affective Disorders, 206, 300–304. https://doi.org/10.1016/j.jad.2016.09.008
Voleti, B., Navarria, A., Liu, R.-J., Banasr, M., Li, N., Terwilliger, R., Sanacora, G., Eid, T., Aghajanian, G., & Duman, R. S. (2013). Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biological Psychiatry, 74(10), 742–749. https://doi.org/10.1016/j.biopsych.2013.04.025
Vollenweider, F. X., Leenders, K. L., Oye, I., Hell, D., & Angst, J. (1997). Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET). European Neuropsychopharmacology: The Journal of the European College of Neuropsychopharmacology, 7(1), 25–38. https://doi.org/10.1016/s0924-977x(96)00042-9
Vollenweider, F. X., Leenders, K. L., Scharfetter, C., Antonini, A., Maguire, P., Missimer, J., & Angst, J. (1997). Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). European Neuropsychopharmacology: The Journal of the European College of Neuropsychopharmacology, 7(1), 9–24. https://doi.org/10.1016/s0924-977x(96)00039-9
Vollmayr, B., & Gass, P. (2013). Learned helplessness: Unique features and translational value of a cognitive depression model. Cell and Tissue Research, 354(1), 171–178. https://doi.org/10.1007/s00441-013-1654-2
Vrieze, E., Pizzagalli, D. A., Demyttenaere, K., Hompes, T., Sienaert, P., de Boer, P., Schmidt, M., & Claes, S. (2013). Reduced reward learning predicts outcome in major depressive disorder. Biological Psychiatry, 73(7), 639–645. https://doi.org/10.1016/j.biopsych.2012.10.014
Vyklicky, V., Korinek, M., Smejkalova, T., Balik, A., Krausova, B., Kaniakova, M., Lichnerova, K., Cerny, J., Krusek, J., Dittert, I., Horak, M., & Vyklicky, L. (2014). Structure, Function, and Pharmacology of NMDA Receptor Channels. Physiological Research, 63, 13.
Walker, A. J., Foley, B. M., Sutor, S. L., McGillivray, J. A., Frye, M. A., & Tye, S. J. (2015). Peripheral proinflammatory markers associated with ketamine response in a preclinical model of antidepressant-resistance. Behavioural Brain Research, 293, 198–202. https://doi.org/10.1016/j.bbr.2015.07.026
Walker, A. K., Budac, D. P., Bisulco, S., Lee, A. W., Smith, R. A., Beenders, B., Kelley, K. W., & Dantzer, R. (2013). NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 38(9), 1609–1616. https://doi.org/10.1038/npp.2013.71
Wan, Y.-Q., Feng, J.-G., Li, M., Wang, M.-Z., Liu, L., Liu, X., Duan, X.-X., Zhang, C.-X., & Wang, X.-B. (2018). Prefrontal cortex miR-29b-3p plays a key role in the antidepressant-like effect of ketamine in rats. Experimental & Molecular Medicine, 50(10), 1–14. https://doi.org/10.1038/s12276-018-0164-4
Wang, C., Zheng, D., Xu, J., Lam, W., & Yew, D. T. (2013). Brain damages in ketamine addicts as revealed by magnetic resonance imaging. Frontiers in Neuroanatomy, 7, 23. https://doi.org/10.3389/fnana.2013.00023
Wang, H.-X., & Gao, W.-J. (2009). Cell Type-Specific Development of NMDA Receptors in the Interneurons of Rat Prefrontal Cortex. Neuropsychopharmacology, 34(8), 2028–2040. https://doi.org/10.1038/npp.2009.20
Wang, N., Yu, H.-Y., Shen, X.-F., Gao, Z.-Q., Yang, C., Yang, J.-J., & Zhang, G.-F. (2015). The rapid antidepressant effect of ketamine in rats is associated with down-regulation of pro-inflammatory cytokines in the hippocampus. Upsala Journal of Medical Sciences, 120(4), 241–248. https://doi.org/10.3109/03009734.2015.1060281
Wang, X., Yang, Y., Zhou, X., Wu, J., Li, J., Jiang, X., Qu, Q., Ou, C., Liu, L., & Zhou, S. (2011). Propofol pretreatment increases antidepressant-like effects induced by acute administration of ketamine in rats receiving forced swimming test. Psychiatry Research, 185(1–2), 248–253. https://doi.org/10.1016/j.psychres.2010.04.046
Wang, Y., Chiou, A. L., Yang, S. T., & Lin, J. C. (1995). Ketamine antagonizes hypoxia-induced dopamine release in rat striatum. Brain Research, 693(1–2), 233–245. https://doi.org/10.1016/0006-8993(95)00758-i
Weathers, F. W., Blake, D. D., Schnurr, P. P., Kaloupek, D. G., Marx, B. P., & Keane, T. M. (n.d.). The Clinician-Administered PTSD Scale for DSM-5 (CAPS-5)—Past Month. National Center for PTSD. https://www.ptsd.va.gov/professional/assessment/documents/CAPS_5_Past_Month.pdf
Weathers, Frank W., Bovin, M. J., Lee, D. J., Sloan, D. M., Schnurr, P. P., Kaloupek, D. G., Keane, T. M., & Marx, B. P. (2018). The Clinician-Administered PTSD Scale for DSM-5 (CAPS-5): Development and initial psychometric evaluation in military veterans. Psychological Assessment, 30(3), 383–395. https://doi.org/10.1037/pas0000486
Weber, M. M., & Emrich, H. M. (1988). Current and historical concepts of opiate treatment in psychiatric disorders. International Clinical Psychopharmacology, 3(3), 255–266. https://doi.org/10.1097/00004850-198807000-00007
Wheeler, D., Boutelle, M. G., & Fillenz, M. (1995). The role of N-methyl-D-aspartate receptors in the regulation of physiologically released dopamine. Neuroscience, 65(3), 767–774. https://doi.org/10.1016/0306-4522(95)93905-7
White, P. F., Ham, J., Way, W. L., & Trevor, A. J. (1980). Pharmacology of ketamine isomers in surgical patients. Anesthesiology, 52(3), 231–239. https://doi.org/10.1097/00000542-198003000-00008
White, P. F., Schüttler, J., Shafer, A., Stanski, D. R., Horai, Y., & Trevor, A. J. (1985). Comparative pharmacology of the ketamine isomers. Studies in volunteers. British Journal of Anaesthesia, 57(2), 197–203. https://doi.org/10.1093/bja/57.2.197
Wichers, M. C., Koek, G. H., Robaeys, G., Verkerk, R., Scharpé, S., & Maes, M. (2005). IDO and interferon-alpha-induced depressive symptoms: A shift in hypothesis from tryptophan depletion to neurotoxicity. Molecular Psychiatry, 10(6), 538–544. https://doi.org/10.1038/sj.mp.4001600
Widman, A. J., & McMahon, L. L. (2018). Disinhibition of CA1 pyramidal cells by low-dose ketamine and other antagonists with rapid antidepressant efficacy. Proceedings of the National Academy of Sciences, 115(13), E3007–E3016. https://doi.org/10.1073/pnas.1718883115
Wilkinson, S. T., Ballard, E. D., Bloch, M. H., Mathew, S. J., Murrough, J. W., Feder, A., Sos, P., Wang, G., Zarate, C. A., & Sanacora, G. (2018). The Effect of a Single Dose of Intravenous Ketamine on Suicidal Ideation: A Systematic Review and Individual Participant Data Meta-Analysis. The American Journal of Psychiatry, 175(2), 150–158. https://doi.org/10.1176/appi.ajp.2017.17040472
Williams, N. R., Heifets, B. D., Blasey, C., Sudheimer, K., Pannu, J., Pankow, H., Hawkins, J., Birnbaum, J., Lyons, D. M., Rodriguez, C. I., & Schatzberg, A. F. (2018). Attenuation of Antidepressant Effects of Ketamine by Opioid Receptor Antagonism. American Journal of Psychiatry, 175(12), 1205–1215. https://doi.org/10.1176/appi.ajp.2018.18020138
Willner, P. (2017). The chronic mild stress (CMS) model of depression: History, evaluation and usage. Neurobiology of Stress, 6, 78–93. https://doi.org/10.1016/j.ynstr.2016.08.002
Wilson, J. A., Nimmo, A. F., Fleetwood-Walker, S. M., & Colvin, L. A. (2008). A randomised double blind trial of the effect of pre-emptive epidural ketamine on persistent pain after lower limb amputation. Pain, 135(1–2), 108–118. https://doi.org/10.1016/j.pain.2007.05.011
Winstock, A. R., Mitcheson, L., Gillatt, D. A., & Cottrell, A. M. (2012). The prevalence and natural history of urinary symptoms among recreational ketamine users. BJU International, 110(11), 1762–1766. https://doi.org/10.1111/j.1464-410X.2012.11028.x
Winter, H., & Irle, E. (2004). Hippocampal volume in adult burn patients with and without posttraumatic stress disorder. The American Journal of Psychiatry, 161(12), 2194–2200. https://doi.org/10.1176/appi.ajp.161.12.2194
Witkin, J. M., Monn, J. A., Schoepp, D. D., Li, X., Overshiner, C., Mitchell, S. N., Carter, G., Johnson, B., Rasmussen, K., & Rorick-Kehn, L. M. (2016). The Rapidly Acting Antidepressant Ketamine and the mGlu2/3 Receptor Antagonist LY341495 Rapidly Engage Dopaminergic Mood Circuits. The Journal of Pharmacology and Experimental Therapeutics, 358(1), 71–82. https://doi.org/10.1124/jpet.116.233627
Wohleb, E. S., Wu, M., Gerhard, D. M., Taylor, S. R., Picciotto, M. R., Alreja, M., & Duman, R. S. (2016). GABA interneurons mediate the rapid antidepressant-like effects of scopolamine. The Journal of Clinical Investigation, 126(7), 2482–2494. https://doi.org/10.1172/JCI85033
Wray, N. H., Schappi, J. M., Singh, H., Senese, N. B., & Rasenick, M. M. (2019). NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Molecular Psychiatry, 24(12), 1833–1843. https://doi.org/10.1038/s41380-018-0083-8
Xiong, Z., Zhang, K., Ishima, T., Ren, Q., Ma, M., Pu, Y., Chang, L., Chen, J., & Hashimoto, K. (2019). Lack of rapid antidepressant effects of Kir4.1 channel inhibitors in a chronic social defeat stress model: Comparison with (R)-ketamine. Pharmacology, Biochemistry, and Behavior, 176, 57–62. https://doi.org/10.1016/j.pbb.2018.11.010
Xiong, Z., Zhang, K., Ren, Q., Chang, L., Chen, J., & Hashimoto, K. (2019). Increased expression of inwardly rectifying Kir4.1 channel in the parietal cortex from patients with major depressive disorder. Journal of Affective Disorders, 245, 265–269. https://doi.org/10.1016/j.jad.2018.11.016
Xu, K., Krystal, J. H., Ning, Y., Chen, D. C., He, H., Wang, D., Ke, X., Zhang, X., Ding, Y., Liu, Y., Gueorguieva, R., Wang, Z., Limoncelli, D., Pietrzak, R. H., Petrakis, I. L., Zhang, X., & Fan, N. (2015). Preliminary analysis of positive and negative syndrome scale in ketamine-associated psychosis in comparison with schizophrenia. Journal of Psychiatric Research, 61, 64–72. https://doi.org/10.1016/j.jpsychires.2014.12.012
Xu, Y., Hackett, M., Carter, G., Loo, C., Gálvez, V., Glozier, N., Glue, P., Lapidus, K., McGirr, A., Somogyi, A. A., Mitchell, P. B., & Rodgers, A. (2016). Effects of Low-Dose and Very Low-Dose Ketamine among Patients with Major Depression: A Systematic Review and Meta-Analysis. The International Journal of Neuropsychopharmacology, 19(4). https://doi.org/10.1093/ijnp/pyv124
Xue, J.-G., Masuoka, T., Gong, X.-D., Chen, K.-S., Yanagawa, Y., Law, S. K. A., & Konishi, S. (2011). NMDA receptor activation enhances inhibitory GABAergic transmission onto hippocampal pyramidal neurons via presynaptic and postsynaptic mechanisms. Journal of Neurophysiology, 105(6), 2897–2906. https://doi.org/10.1152/jn.00287.2010
Yamaguchi, J.-I., Toki, H., Qu, Y., Yang, C., Koike, H., Hashimoto, K., Mizuno-Yasuhira, A., & Chaki, S. (2018). (2R,6R)-Hydroxynorketamine is not essential for the antidepressant actions of (R)-ketamine in mice. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 43(9), 1900–1907. https://doi.org/10.1038/s41386-018-0084-y
Yamanaka, H., Yokoyama, C., Mizuma, H., Kurai, S., Finnema, S. J., Halldin, C., Doi, H., & Onoe, H. (2014). A possible mechanism of the nucleus accumbens and ventral pallidum 5-HT1B receptors underlying the antidepressant action of ketamine: A PET study with macaques. Translational Psychiatry, 4, e342. https://doi.org/10.1038/tp.2013.112
Yanagihara, Y., Ohtani, M., Kariya, S., Uchino, K., Hiraishi, T., Ashizawa, N., Aoyama, T., Yamamura, Y., Yamada, Y., & Iga, T. (2003). Plasma concentration profiles of ketamine and norketamine after administration of various ketamine preparations to healthy Japanese volunteers. Biopharmaceutics & Drug Disposition, 24(1), 37–43. https://doi.org/10.1002/bdd.336
Yang, B., Ren, Q., Ma, M., Chen, Q.-X., & Hashimoto, K. (2016). Antidepressant Effects of (+)-MK-801 and (-)-MK-801 in the Social Defeat Stress Model. The International Journal of Neuropsychopharmacology, 19(12). https://doi.org/10.1093/ijnp/pyw080
Yang, B., Zhang, J.-C., Han, M., Yao, W., Yang, C., Ren, Q., Ma, M., Chen, Q.-X., & Hashimoto, K. (2016). Comparison of R-ketamine and rapastinel antidepressant effects in the social defeat stress model of depression. Psychopharmacology, 233(19–20), 3647–3657. https://doi.org/10.1007/s00213-016-4399-2
Yang, C, Shirayama, Y., Zhang, J., Ren, Q., Yao, W., Ma, M., Dong, C., & Hashimoto, K. (2015). R-ketamine: A rapid-onset and sustained antidepressant without psychotomimetic side effects. Translational Psychiatry, 5(9), e632–e632. https://doi.org/10.1038/tp.2015.136
Yang, Chun, Han, M., Zhang, J., Ren, Q., & Hashimoto, K. (2016). Loss of parvalbumin-immunoreactivity in mouse brain regions after repeated intermittent administration of esketamine, but not R-ketamine. Psychiatry Research, 239, 281–283. https://doi.org/10.1016/j.psychres.2016.03.034
Yang, Chun, Hong, T., Shen, J., Ding, J., Dai, X.-W., Zhou, Z.-Q., & Yang, J.-J. (2013). Ketamine exerts antidepressant effects and reduces IL-1β and IL-6 levels in rat prefrontal cortex and hippocampus. Experimental and Therapeutic Medicine, 5(4), 1093–1096. https://doi.org/10.3892/etm.2013.930
Yang, Chun, Hu, Y.-M., Zhou, Z.-Q., Zhang, G.-F., & Yang, J.-J. (2013). Acute administration of ketamine in rats increases hippocampal BDNF and mTOR levels during forced swimming test. Upsala Journal of Medical Sciences, 118(1), 3–8. https://doi.org/10.3109/03009734.2012.724118
Yang, Chun, Kobayashi, S., Nakao, K., Dong, C., Han, M., Qu, Y., Ren, Q., Zhang, J.-C., Ma, M., Toki, H., Yamaguchi, J.-I., Chaki, S., Shirayama, Y., Nakazawa, K., Manabe, T., & Hashimoto, K. (2018). AMPA Receptor Activation-Independent Antidepressant Actions of Ketamine Metabolite (S)-Norketamine. Biological Psychiatry, 84(8), 591–600. https://doi.org/10.1016/j.biopsych.2018.05.007
Yang, Chun, Qu, Y., Abe, M., Nozawa, D., Chaki, S., & Hashimoto, K. (2017). (R)-Ketamine Shows Greater Potency and Longer Lasting Antidepressant Effects Than Its Metabolite (2R,6R)-Hydroxynorketamine. Biological Psychiatry, 82(5), e43–e44. https://doi.org/10.1016/j.biopsych.2016.12.020
Yang, Chun, Qu, Y., Fujita, Y., Ren, Q., Ma, M., Dong, C., & Hashimoto, K. (2017). Possible role of the gut microbiota-brain axis in the antidepressant effects of (R)-ketamine in a social defeat stress model. Translational Psychiatry, 7(12), 1294. https://doi.org/10.1038/s41398-017-0031-4
Yang, Chun, Ren, Q., Qu, Y., Zhang, J.-C., Ma, M., Dong, C., & Hashimoto, K. (2018). Mechanistic Target of Rapamycin-Independent Antidepressant Effects of (R)-Ketamine in a Social Defeat Stress Model. Biological Psychiatry, 83(1), 18–28. https://doi.org/10.1016/j.biopsych.2017.05.016
Yang, Chun, Zhou, Z., Gao, Z., Shi, J., & Yang, J.-J. (2013). Acute increases in plasma mammalian target of rapamycin, glycogen synthase kinase-3β, and eukaryotic elongation factor 2 phosphorylation after ketamine treatment in three depressed patients. Biological Psychiatry, 73(12), e35-36. https://doi.org/10.1016/j.biopsych.2012.07.022
Yang, J.-J., Wang, N., Yang, C., Shi, J.-Y., Yu, H.-Y., & Hashimoto, K. (2015). Serum interleukin-6 is a predictive biomarker for ketamine’s antidepressant effect in treatment-resistant patients with major depression. Biological Psychiatry, 77(3), e19–e20. https://doi.org/10.1016/j.biopsych.2014.06.021
Yang, X., Yang, Q., Wang, X., Luo, C., Wan, Y., Li, J., Liu, K., Zhou, M., & Zhang, C. (2014). MicroRNA expression profile and functional analysis reveal that miR-206 is a critical novel gene for the expression of BDNF induced by ketamine. Neuromolecular Medicine, 16(3), 594–605. https://doi.org/10.1007/s12017-014-8312-z
Yang, Y., Cui, Y., Sang, K., Dong, Y., Ni, Z., Ma, S., & Hu, H. (2018). Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature, 554(7692), 317–322. https://doi.org/10.1038/nature25509
Yao, N., Skiteva, O., Zhang, X., Svenningsson, P., & Chergui, K. (2018). Ketamine and its metabolite (2R,6R)-hydroxynorketamine induce lasting alterations in glutamatergic synaptic plasticity in the mesolimbic circuit. Molecular Psychiatry, 23(10), 2066–2077. https://doi.org/10.1038/mp.2017.239
Yasuda, S., Liang, M.-H., Marinova, Z., Yahyavi, A., & Chuang, D.-M. (2009). The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Molecular Psychiatry, 14(1), 51–59. https://doi.org/10.1038/sj.mp.4002099
Yoon, G., Petrakis, I. L., & Krystal, J. H. (2019). Association of Combined Naltrexone and Ketamine With Depressive Symptoms in a Case series of Patients With Depression and Alcohol Use Disorder. JAMA Psychiatry, 76(3), 337–338. https://doi.org/10.1001/jamapsychiatry.2018.3990
Young, R. C., Biggs, J. T., Ziegler, V. E., & Meyer, D. A. (1978). A Rating Scale for Mania: Reliability, Validity and Sensitivity. British Journal of Psychiatry, 133(5), 429–435. https://doi.org/10.1192/bjp.133.5.429
Yovell, Y., Bar, G., Mashiah, M., Baruch, Y., Briskman, I., Asherov, J., Lotan, A., Rigbi, A., & Panksepp, J. (2016). Ultra-Low-Dose Buprenorphine as a Time-Limited Treatment for Severe Suicidal Ideation: A Randomized Controlled Trial. The American Journal of Psychiatry, 173(5), 491–498. https://doi.org/10.1176/appi.ajp.2015.15040535
Yuen, E. Y., Wei, J., Liu, W., Zhong, P., Li, X., & Yan, Z. (2012). Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron, 73(5), 962–977. https://doi.org/10.1016/j.neuron.2011.12.033
Zanos, P., Highland, J. N., Liu, X., Troppoli, T. A., Georgiou, P., Lovett, J., Morris, P. J., Stewart, B. W., Thomas, C. J., Thompson, S. M., Moaddel, R., & Gould, T. D. (2019). (R)-Ketamine exerts antidepressant actions partly via conversion to (2R,6R)-hydroxynorketamine, while causing adverse effects at sub-anaesthetic doses. British Journal of Pharmacology, 176(14), 2573–2592. https://doi.org/10.1111/bph.14683
Zanos, P., Moaddel, R., Morris, P. J., Georgiou, P., Fischell, J., Elmer, G. I., Alkondon, M., Yuan, P., Pribut, H. J., Singh, N. S., Dossou, K. S. S., Fang, Y., Huang, X.-P., Mayo, C. L., Albuquerque, E. X., Thompson, S. M., Thomas, C. J., Zarate, C. A., & Gould, T. D. (2017). Zanos et al. Reply. Nature, 546(7659), E4–E5. https://doi.org/10.1038/nature22085
Zanos, P., Moaddel, R., Morris, P. J., Georgiou, P., Fischell, J., Elmer, G. I., Alkondon, M., Yuan, P., Pribut, H. J., Singh, N. S., Dossou, K. S. S., Fang, Y., Huang, X.-P., Mayo, C. L., Wainer, I. W., Albuquerque, E. X., Thompson, S. M., Thomas, C. J., Zarate, C. A., & Gould, T. D. (2016). NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature, 533(7604), 481–486. https://doi.org/10.1038/nature17998
Zanos, P., Moaddel, R., Morris, P. J., Riggs, L. M., Highland, J. N., Georgiou, P., Pereira, E. F. R., Albuquerque, E. X., Thomas, C. J., Zarate, C. A., & Gould, T. D. (2018). Ketamine and Ketamine Metabolite Pharmacology: Insights into Therapeutic Mechanisms. Pharmacological Reviews, 70(3), 621–660. https://doi.org/10.1124/pr.117.015198
Zanos, P., Nelson, M. E., Highland, J. N., Krimmel, S. R., Georgiou, P., Gould, T. D., & Thompson, S. M. (2017). A Negative Allosteric Modulator for α5 Subunit-Containing GABA Receptors Exerts a Rapid and Persistent Antidepressant-Like Action without the Side Effects of the NMDA Receptor Antagonist Ketamine in Mice. ENeuro, 4(1). https://doi.org/10.1523/ENEURO.0285-16.2017
Zarate, C. A., Brutsche, N. E., Ibrahim, L., Franco-Chaves, J., Diazgranados, N., Cravchik, A., Selter, J., Marquardt, C. A., Liberty, V., & Luckenbaugh, D. A. (2012). Replication of ketamine’s antidepressant efficacy in bipolar depression: A randomized controlled add-on trial. Biological Psychiatry, 71(11), 939–946. https://doi.org/10.1016/j.biopsych.2011.12.010
Zarate, C. A., Brutsche, N., Laje, G., Luckenbaugh, D. A., Venkata, S. L. V., Ramamoorthy, A., Moaddel, R., & Wainer, I. W. (2012). Relationship of ketamine’s plasma metabolites with response, diagnosis, and side effects in major depression. Biological Psychiatry, 72(4), 331–338. https://doi.org/10.1016/j.biopsych.2012.03.004
Zarate, C. A., Mathews, D., Ibrahim, L., Chaves, J. F., Marquardt, C., Ukoh, I., Jolkovsky, L., Brutsche, N. E., Smith, M. A., & Luckenbaugh, D. A. (2013). A Randomized Trial of a Low-Trapping Nonselective N-Methyl-D-Aspartate Channel Blocker in Major Depression. Biological Psychiatry, 74(4), 257–264. https://doi.org/10.1016/j.biopsych.2012.10.019
Zarate, C. A., Singh, J. B., Carlson, P. J., Brutsche, N. E., Ameli, R., Luckenbaugh, D. A., Charney, D. S., & Manji, H. K. (2006). A Randomized Trial of an N-methyl-D-aspartate Antagonist in Treatment-Resistant Major Depression. Archives of General Psychiatry, 63(8), 856. https://doi.org/10.1001/archpsyc.63.8.856
Zarate, C. A., Singh, J. B., Quiroz, J. A., De Jesus, G., Denicoff, K. K., Luckenbaugh, D. A., Manji, H. K., & Charney, D. S. (2006). A double-blind, placebo-controlled study of memantine in the treatment of major depression. The American Journal of Psychiatry, 163(1), 153–155. https://doi.org/10.1176/appi.ajp.163.1.153
Zhang, G.-F., Liu, W.-X., Qiu, L.-L., Guo, J., Wang, X.-M., Sun, H.-L., Yang, J.-J., & Zhou, Z.-Q. (2015). Repeated ketamine administration redeems the time lag for citalopram’s antidepressant-like effects. European Psychiatry: The Journal of the Association of European Psychiatrists, 30(4), 504–510. https://doi.org/10.1016/j.eurpsy.2014.11.007
Zhang, Guang-Fen, Wang, N., Shi, J.-Y., Xu, S.-X., Li, X.-M., Ji, M.-H., Zuo, Z.-Y., Zhou, Z.-Q., & Yang, J.-J. (2013). Inhibition of the L-arginine-nitric oxide pathway mediates the antidepressant effects of ketamine in rats in the forced swimming test. Pharmacology, Biochemistry, and Behavior, 110, 8–12. https://doi.org/10.1016/j.pbb.2013.05.010
Zhang, J., Yao, W., Dong, C., Yang, C., Ren, Q., Ma, M., Han, M., & Hashimoto, K. (2015). Comparison of ketamine, 7,8-dihydroxyflavone, and ANA-12 antidepressant effects in the social defeat stress model of depression. Psychopharmacology, 232(23), 4325–4335. https://doi.org/10.1007/s00213-015-4062-3
Zhang, J.-C., Yao, W., Dong, C., Yang, C., Ren, Q., Ma, M., & Hashimoto, K. (2017). Blockade of interleukin-6 receptor in the periphery promotes rapid and sustained antidepressant actions: A possible role of gut-microbiota-brain axis. Translational Psychiatry, 7(5), e1138. https://doi.org/10.1038/tp.2017.112
Zhang, Ji-Chun, Li, S.-X., & Hashimoto, K. (2014). R (-)-ketamine shows greater potency and longer lasting antidepressant effects than S (+)-ketamine. Pharmacology, Biochemistry, and Behavior, 116, 137–141. https://doi.org/10.1016/j.pbb.2013.11.033
Zhang, Kai, Fujita, Y., & Hashimoto, K. (2018). Lack of metabolism in (R)-ketamine’s antidepressant actions in a chronic social defeat stress model. Scientific Reports, 8(1), 4007. https://doi.org/10.1038/s41598-018-22449-9
Zhang, Kai, & Hashimoto, K. (2019). Lack of Opioid System in the Antidepressant Actions of Ketamine. Biological Psychiatry, 85(6), e25–e27. https://doi.org/10.1016/j.biopsych.2018.11.006
Zhang, Kai, Ma, M., Dong, C., & Hashimoto, K. (2018). Role of Inflammatory Bone Markers in the Antidepressant Actions of (R)-Ketamine in a Chronic Social Defeat Stress Model. The International Journal of Neuropsychopharmacology, 21(11), 1025–1030. https://doi.org/10.1093/ijnp/pyy065
Zhang, Kai, Toki, H., Fujita, Y., Ma, M., Chang, L., Qu, Y., Harada, S., Nemoto, T., Mizuno-Yasuhira, A., Yamaguchi, J.-I., Chaki, S., & Hashimoto, K. (2018). Lack of deuterium isotope effects in the antidepressant effects of (R)-ketamine in a chronic social defeat stress model. Psychopharmacology, 235(11), 3177–3185. https://doi.org/10.1007/s00213-018-5017-2
Zhang, Ke, Xu, T., Yuan, Z., Wei, Z., Yamaki, V. N., Huang, M., Huganir, R. L., & Cai, X. (2016). Essential roles of AMPA receptor GluA1 phosphorylation and presynaptic HCN channels in fast-acting antidepressant responses of ketamine. Science Signaling, 9(458), ra123. https://doi.org/10.1126/scisignal.aai7884
Zhao, X., Venkata, S. L. V., Moaddel, R., Luckenbaugh, D. A., Brutsche, N. E., Ibrahim, L., Zarate, C. A., Mager, D. E., & Wainer, I. W. (2012). Simultaneous population pharmacokinetic modelling of ketamine and three major metabolites in patients with treatment-resistant bipolar depression. British Journal of Clinical Pharmacology, 74(2), 304–314. https://doi.org/10.1111/j.1365-2125.2012.04198.x
Zheng, P., Zeng, B., Zhou, C., Liu, M., Fang, Z., Xu, X., Zeng, L., Chen, J., Fan, S., Du, X., Zhang, X., Yang, D., Yang, Y., Meng, H., Li, W., Melgiri, N. D., Licinio, J., Wei, H., & Xie, P. (2016). Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Molecular Psychiatry, 21(6), 786–796. https://doi.org/10.1038/mp.2016.44
Zhou, W., Wang, N., Yang, C., Li, X.-M., Zhou, Z.-Q., & Yang, J.-J. (2014). Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. European Psychiatry: The Journal of the Association of European Psychiatrists, 29(7), 419–423. https://doi.org/10.1016/j.eurpsy.2013.10.005
Zhou, X., Chen, Z., Yun, W., Ren, J., Li, C., & Wang, H. (2015). Extrasynaptic NMDA Receptor in Excitotoxicity: Function Revisited. The Neuroscientist: A Review Journal Bringing Neurobiology, Neurology and Psychiatry, 21(4), 337–344. https://doi.org/10.1177/1073858414548724
Zhu, Y., Shanley, M., & Ku, S. (2018). HCN channel inhibitor induces a rapid and sustained reversal of social defeat in a chronic social defeat stress model. Neuropsychopharmacology, 43(S1), 77–227. https://doi.org/10.1038/s41386-018-0266-7
Zorzela, L., Golder, S., Liu, Y., Pilkington, K., Hartling, L., Joffe, A., Loke, Y., & Vohra, S. (2014). Quality of reporting in systematic reviews of adverse events: Systematic review. BMJ, 348, f7668–f7668. https://doi.org/10.1136/bmj.f7668
Zuo, D.-Y., Wu, Y.-L., Yao, W.-X., Cao, Y., Wu, C.-F., & Tanaka, M. (2007). Effect of MK-801 and ketamine on hydroxyl radical generation in the posterior cingulate and retrosplenial cortex of free-moving mice, as determined by in vivo microdialysis. Pharmacology, Biochemistry, and Behavior, 86(1), 1–7. https://doi.org/10.1016/j.pbb.2006.05.010